
Recent advances in platinum-group-metal based electrocatalysts for alkaline hydrogen oxidation reaction

Abstract
The reaction kinetics of hydrogen oxidation reactions (HOR) unfavorably decreases by 2~3 orders of magnitude under alkaline conditions, even on the most active platinum-group-metal (PGM) electrocatalysts. This sticky problem severely restricts the efficiency and commercialization of anion-exchange membrane fuel cells (AEMFCs). So far, no other material has HOR electrocatalytic performance comparable to PGM-based electrocatalysts. Forced by the scarce reserves and high prices of PGMs, it is significant to elaborately design and synthesize PGM-based electrocatalysts with ultimately atomic utilization and substantially improved alkaline HOR performance. In this review, we summarize recent advances in the structure engineering approaches to synthesis of advanced PGM-based nanocatalysts toward enhanced alkaline HOR performance. The generally acknowledged catalytic mechanisms with corresponding activity descriptors are reviewed firstly to deeply understand the discrepancies in the HOR kinetics of alkaline and acidic reactions. Then, several representative strategies are emphasized and discussed at length by changing the chemical and coordination environment and size/morphology of nanocatalysts. Meanwhile, the influence factors for the performance of AEMFC devices constructed by PGM-based anode catalysts are briefly highlighted. In conclusion, strategies for boosting the electrocatalytic performance and challenges on the roles of catalytic mechanism insights and practical AEMFC applications are finally outlined. We hope this review will guide the design and catalytic mechanism research of novel PGM-based alkaline HOR catalysts, thereby promoting their further development and application in AEMFC technologies.
Keywords
INTRODUCTION
Hydrogen (H2), as an emerging zero-carbon energy carrier, has shown superior advantages over traditional fossil energy in terms of ecological friendliness and sustainable development, which encourages the burgeoning of advanced and economically feasible hydrogen conversion technologies[1]. The most promising conversion devices for the consumption and utilization of hydrogen are low-temperature ion-exchange membrane hydrogen fuel cells, i.e., the proton-exchange membrane fuel cells (PEMFCs) and anion-exchange membrane fuel cells (AEMFCs)[2-5]. For PEMFCs, the heavy demands for expensive perfluorinated sulfonic acid membranes and precious metal materials in strong acidic corrosive operating conditions seriously restrict their large-scale development and application[6,7]. In contrast, AEMFCs have gained increasing attention because of their less corrosive alkaline nature that permits the use of cost-effective hydrocarbon-based membranes and metal materials[8-11]. It has been reported that there are a number of non-precious-metal nanomaterials exhibiting excellent electrocatalytic performance for cathodic oxygen reduction reactions (ORR) in alkaline media, some of which are even comparable to the platinum-group-metal (PGM) nanomaterials[12-14]. Unfortunately, the anodic hydrogen oxidation reaction (HOR) in alkaline media delivers much slower intrinsic kinetics than that in acidic media. Even for state-of-the-art Pt, the exchange current density presents a decline of 2~3 orders of magnitude, which immensely retards the output performances of AEMFCs[15,16]. To get insight into the reaction kinetics of alkaline HOR and guide the formulation of high-efficiency catalysts, researchers have put forward several plausible mechanisms[17-19], commonly including (1) the hydrogen binding energy (HBE) theory, where the binding ability of adsorbed H (Had) dominantly affects the Volmer step (the oxidative removal of Had: Had + OH− → H2O + e−) of alkaline HOR; (2) the bifunctional mechanism, in which the hydroxyl binding energy (OHBE) is considered as another indispensable parameter affecting the oxidative removal of Had; (3) the potential of zero free charge (PZFC) theory, i.e., the distance between PZFC and hydrogen equilibrium potential impacts the transfer rate of OH− through the interface electric double layer by regulating the recombination energy of interfacial water. In addition, several other related theories, such as the effects of solvent molecules, anions and cations, and quasi-characteristic adsorption, have also been proposed[11]. These theories indicate that the intrinsic feature of a catalyst and the solution phase both play indispensable roles in the electrocatalytic reaction kinetics, resulting in the complexity and controversy of alkaline HOR mechanisms.
Generally, HBE theory is mainly related to the thermodynamical Gibbs free energy for hydrogen (ΔGH). Based on the Sabatier principle, an excellent catalyst surface needs to maintain moderate binding energy with the reaction intermediate. If the binding energy is too weak, the reactant will have difficulty combining with the catalyst to form an adsorbed intermediate, whereas a much stronger binding energy will make it difficult for the intermediate to further form products and detach from the catalyst surface. Thus, PGM catalysts with their ΔGH located near 0 eV should exhibit the most suitable adsorption strength for Had intermediates, thereby exhibiting excellent HOR electrocatalytic performance[20,21]. However, given the low reserves and high prices of PGMs, feasible programs must be taken to improve the cost-effectiveness while increasing their electrocatalytic activities. Strategies for improving activities are roughly divided into two aspects, i.e., augmenting the number of catalytic active sites and optimizing the intrinsic activity of each active site[22]. Typically, regulating the size and dimension of PGMs to obtain nanometer-level and low-dimensional structures is beneficial for exposing abundant active sites[23-26]. Introducing earth-abundant non-PGMs or non-metallic elements into PGMs to form multicomponent structures can help regulate the electronic densities of their active sites[27-29]. Precise control of the hetero-atomic arrangements can affect the ensemble effect, ligand effect, and strain effect, thereby further improving the intrinsic activities of active sites[30,31]. Tailoring the morphologies, compositions, and atomic structures of PGM-based nanocatalysts enables the modulation of electron structures, which effectively regulates the binding energy of surface reactive atoms for reaction intermediates. Ideally, these strategies are not mutually exclusive and can be applied simultaneously, giving rise to maximized improvement of activity. In terms of alkaline HOR electrocatalysis, experimental conditions and different reaction intermediates should be fully considered during operation, which makes the rational design of PGM-based nanocatalysts with excellent electrocatalytic performance still an imperative prerequisite and far from expectations[16,19,32]. To this end, a systematic investigation on exploring the inherent relationship between the structure and electrocatalytic performance of PGM-based nanocatalysts, which is urgently required for alkaline HOR electrocatalysis, should be implemented.
In this review, we summarize current advances in the research on designing excellent PGM-based nanomaterials toward alkaline HOR electrocatalysis. First, the generally acknowledged catalytic mechanisms with corresponding activity descriptors are introduced to deeply understand the discrepancies in reaction kinetics between alkaline and acidic conditions. Then, guided by the catalytic mechanisms of alkaline HOR, we systematically outline several design strategies to engineer PGM-based catalysts with excellent electrocatalytic performance by modulating the crystal structure, size and atomic dispersion, dimensional, composition, surface active layer, interface, etc. Due to the different electrochemical testing conditions in the laboratory and practical AEMFCs, the catalytic performances might exhibit remarkable differences[33-35]. Thus, the performance evaluation on AEMFC devices assembled with PGM-based catalysts is also discussed. In the end, we emphasize the current challenges in sluggish alkaline HOR kinetics and the practical applications of catalytic materials. We also outline our perspectives on the scalable production of low-PGM catalysts and the commercialization of fuel cells.
ALKALINE HOR FUNDAMENTAL FOR PGM-BASED CATALYSTS
With the development of alkaline polymer electrolyte membrane technology, AEMFCs have become popular alternative equipment for promoting the fuel cell industries in recent years. The sluggish alkaline HOR electrocatalytic process, however, strictly limits the performance of AEMFCs, which sparks in-depth exploration by researchers on the catalytic reaction mechanism of HOR.
Mechanism
The reaction equation and related elementary steps of the alkaline HOR process are described in Equations 1-4, in which the overall reaction process (Equation 1) involves the formation of Had intermediate, followed by the formation of water[18,36].
overall reaction: H2 + 2OH- ⇌ 2H2O + 2e- (1)
Tafel: H2 + 2* ⇌ 2Had (2)
Heyrovsky: H2 + OH-+* ⇄ Had + H2O + e- (3)
Volmer: Had + OH- ⇄ H2O + e- + * (4)
Specifically, there are two ways to produce the Had intermediates. In one way, an H2 molecular is chemically absorbed on surface active sites (*) and dissociated into two absorbed Had without electron transfer (Equation 2). Alternatively, the H2 molecular combinates with adjacent OH− to generate an absorbed Had intermediate and H2O molecular through electrochemical dissociation, coupled with liberating an electron (Equation 3). As for the oxidative desorption of Had, the above formed Had intermediate further reacts with adjacent OH− to generate H2O molecular, simultaneously to liberate an electron and an active site
According to the three elementary steps, the adsorption and desorption of Had intermediates on PGM-based catalysts run through the entire reaction process, which suggests the indispensable role of HBE. Both Heyrovsky and Volmer steps involve the participation of OH- and H2O, which means the reaction kinetics are also closely related to the energy barriers for binding OH- and forming water[41,46]. Meanwhile, the kinetics of electrode reaction processes are yet subject to the microenvironment of electrolytes involving the electric double layer, water structure, ion effects, etc. So, it is difficult to conclude a universal mechanism of alkaline HOR under the controversy of theories. Nevertheless, some activity descriptors have been proposed and widely validated by decades of extensive efforts, providing significant assistance in guiding the design of high-performance PGM-based catalysts.
HBE theory
It is worth noting that the Had intermediate is closely linked to the reaction path, whereas the source and existing form of OH– reacting with Had during the electrocatalytic process is still highly controversial[18]. The HBE theory suggests that Had directly reacts with OH− in electrolytes to generate H2O[20]. This theory refers to the influence of corresponding strength of the metal-hydrogen (M-H) bonds on the HOR rate, which is usually represented by Gibbs free energy in thermodynamics (ΔGH)[21]. In early 1972, Trasatti first proposed using HBE to describe the acidic hydrogen evolution reaction (HER, the inverse process of HOR) and HOR performances[47]. They found that the HER/HOR exchange current densities of many metal catalysts exhibit typical volcano-like curve relationships with the strength of their M-H bonds. Then, in 2005, with the development of theoretical calculation chemistry, Norskov et al. concluded a similar volcano-like relationship between acidic HER/HOR catalytic activities and ΔGH by employing DFT calculations

Figure 1. (A) The relationships between acidic HER/HOR exchange current densities of different metals and the chemisorption energy of hydrogen (ΔEH, top), or the adsorption free energy of hydrogen obtained by kinetic simulation (ΔGH, bottom). Reproduced with permission from[21]. Copyright 2005, IOP Publishing; (B) Alkaline HER exchange current densities of monometallic surfaces plotted as a function of the calculated HBEs. Reproduced with permission from[49]. Copyright 2013, Royal Society of Chemistry; (C) Steady-state CVs of Pt; (D) HBEs on Pt(110) and Pt(100) surfaces obtained from (C) as a function of solution pH; (E) Overpotentials of the HOR/HER on Pt(110) and Pt(100) as a function of HBEs. Reproduced with permission from[50]. Copyright 2015, Springer Nature.
Given the varieties of HBE, Sheng et al. continued to explore the relationship between HER/HOR catalytic activities and HBE under different pH conditions, with Pt electrodes as the research target[50]. From the electrochemical experiment, they found that the HUPD peaks of Pt(110) and Pt(100) planes obtained by cyclic voltammetry (CV) curves all shift toward more positive potentials as the pH increases and the decreased values of corresponding HBE calculated by the equation of ΔGH = –nFE [Figure 1C and D]. By plotting the overpotential (η) with HBE, it can be seen that the HER/HOR activities of Pt(110) and Pt(100) planes are negatively correlated with the strength of HBE [Figure 1E]. They finally concluded that the OH– in alkaline media affects the strength of HBE, thereby affecting the catalytic activity of hydrogen electrodes. To gain insight into the reasons for the significantly poorer alkaline catalytic performance, their groups further studied the relevance of HER/HOR activities of other PGM catalysts, the pH of electrolytes, and HBE[39]. The experimental results show that the potentials of HUPD peaks in Pt/C, Ir/C, Pd/C, and Rh/C catalysts exhibit a significant positive correlation with the pH, and their exchange current densities are also related to the calculated HBE from the HUPD peak, further proving that HBE should be the main active descriptor of hydrogen electrodes. As for the adsorption of OH–, they conducted CO stripping experiments to explore the adsorption process and found that the initial potential of OH adsorption decreases with the increased pH[39,51]. That is to say, the strengthened oxophilicity instead corresponds to the inferior catalytic performance, suggesting that the oxophilicity of PGM catalysts may have little effect on the HOR activity.
The above conclusions indicate that the HBE of PGM catalysts calculated from the potentials of the HUPD peak is linearly correlated with the pH. However, some special catalysts remain controversial. For instance, the calculated HBE of Pt (111) single crystal electrodes does not alter with pH variation[52]. On this basis, HBE theory was revised and a new activity descriptor of the apparent HBE ( HBEapp) was proposed in 2018[53,54]. HBEapp represents the co-presence of Had and H2Oad intermediates on the surface of metal catalysts, which is thermodynamically expressed as
OHBE theory
According to the elementary steps of alkaline HOR electrocatalysis, OH− exists in both the Heyrovsky and Volmer steps, which means that the adsorption ability of catalysts for OH species may be another significant factor in effecting the catalytic kinetics. Although the form of OH species participating in the elementary steps is still controversial, there are many explorations that have confirmed that the OHBE of catalysts is closely related to the alkaline HOR electrocatalytic activity[55-61]. Thus, a bi-functional mechanism containing both the HBE and OHBE theories has been widely proposed to explain the alkaline HOR kinetics. Besides considering the Had intermediates, OHBE theory acknowledges the adsorbed OH species on the surface active sites (OHad) as the reaction intermediates as well. In the year of 2011, Subbaraman et al. first depicted the OHBE theory based on the alkaline HER electrochemical experiments on the Pt electrodes modified with Ni(OH)2 nanoclusters[62]. They chose several Pt electrodes, including the Pt(111) and Pt(110) single crystal electrodes and and Pt/C nanoparticles (NPs). The electrochemical test results show that Ni(OH)2 nanocluster-modified Pt electrodes all deliver significant improvement of activities compared to their counterparts. The modified Ni(OH)2 should promote the dissociation process of H2O in the alkaline medium and, thus, accelerate the reaction kinetics. Based on the research, they proposed a bi-functional mechanism for alkaline HER electrocatalysis, where H2O dissociates into OHad and Had intermediates adsorbed on the oxyphilic [Ni(OH)2] sites and Pt sites, respectively. The catalytic mechanism process is shown in [Figure 2A]. Moreover, their group further prepared a series of Ni(OH)2-modified transition metal catalysts toward catalyzing HER in alkaline and acidic media to ascertain whether the bi-functional mechanism is universally applicable [Figure 2B][63]. For alkaline HER electrocatalysis, modifying Ni(OH)2 functional groups on transition metals can indeed significantly improve their activities, demonstrating the advantageous role of oxyphilic species on reaction kinetics. Notably, the difference in performance between Ir and Pt in acid is much smaller than that in alkaline conditions, whereas this discrepancy can be narrowed after modifying with Ni(OH)2. This indicates that introducing oxyphilic species can promote the dissociation process of H2O and, thus, improve the alkaline HER kinetics. The above conclusion was later proved by testing the electrocatalytic performances of Pt(111) single crystal electrodes modified with four different transition metal (Mn/Fe/Co/Ni) hydroxides[64].

Figure 2. (A) Schematic diagram of alkaline HER electrocatalytic mechanism processes on the Pt electrodes modified with Ni(OH)2 nanoclusters. Reproduced with permission from[62]. Copyright 2011, AAAS; (B) Comparison of HER activities for both bare metal surfaces and Ni(OH)2-modified surfaces in 0.1 M HClO4 and 0.1 M KOH. Reproduced with permission from[63]. Copyright 2012, John Wiley and Sons; (C) HOR/HER polarization curves for Pt(111) and Pt(111) modified with Ni(OH)2; (D) Schematic representation of HOR on Ni(OH)2/Pt(111); (E) HOR/HER polarization curves for Pt, Ir, and PtRu alloys with 50% Ru and 90% Ru. Reproduced with permission from[55]. Copyright 2013, Springer Nature; (F) Plot of the simulated (black line) and experimentally measured (blue circles) rates of HER on Pt(111) and Pt(553) with different adsorbed metal atoms versus hydroxide adsorption energy at 0 VRHE; The orange diamonds correspond to the experimentally measured rate on the Mo* and Re* decorated steps plotted against the DFT calculated potential to oxidize the step. Reproduced with permission from[65]. Copyright 2020, Springer Nature.
In terms of the alkaline HOR electrocatalysis, Strmcnik et al. also confirmed the feasibility of OHBE theory with Ni(OH)2/Pt(111) catalysts [Figure 2C][55]. Specifically, Ni(OH)2 clusters act as oxyphilic active sites that are conducive to the adsorption of reactive OHad, and Pt serves as active sites for Had. The enhanced oxyphilic nature of OHad is beneficial for the oxidative removal of Had and, thus, accelerates the Heyrovsky/Volmer step of alkaline HOR electrocatalysis [Figure 2D]. Moreover, Ir and PtRu alloy catalysts were also used to catalyze alkaline HOR, both of which exhibit excellent performances [Figure 2E]. The abundant oxyphilic sites (such as the Ir defects and Ru atoms) on the catalyst surface encourage the adsorption of OH species, leading to improved electrocatalytic performance. Therefore, OHad is a key intermediate reacting with Had to form H2O during the alkaline HOR electrocatalysis process. In fact, the actual role of OHad is still unclear at that moment, and so does the relationship between OHBE and the hydrogen electrode activity. Through experimental exploration, Mccrum and Koper reported that there is a volcano-like relationship between alkaline HER activity and the strength of OHBE by investigating the Pt(553) single crystal electrodes that selectively deposited different types of metal atoms at the step edges [Figure 2F][65]. They found that the modified Ru atoms can significantly improve the activity of Pt(553) electrodes by 65-fold enhancements. In combination with the DFT calculation, the energy barrier of H2O splitting was calculated to be related to the adsorption strength of OH, which corresponds to the volcano-like curve obtained from experiment results. This work not only confirms the applicability of the bi-functional mechanism for alkaline hydrogen electrode catalysis but also guides the design of high-performance catalysts featured with optimal HBE and OHBE simultaneously.
PZFC theory and other essential factors
HBE and OHBE theories have already been accepted and widely used to elucidate the alkaline hydrogen electrode catalysis according to extensive effort in experiments and theoretical calculations. Meanwhile, another theory regarding the recombination energy influence of interfacial water has also been proposed, i.e., the PZFC theory[66]. This theory explains the reason for the slow kinetics of alkaline HOR/HER electrocatalysis, which was advanced by Ledezma-Yanez et al., who studied the kinetic processes of the adsorption and desorption of hydrogen at different pH levels[66]. PZFC refers to the electrode potential when there is no residual charge on the surface of electrodes. At this moment, there will be no electric double layer caused by residual charges at the interface between the electrode and solution. Under alkaline conditions, it can be measured using the laser-induced temperature jump method. Taking Pt(111) and Pt(111)/Ni(OH)2 as the experimental objects, the introduction of Ni(OH)2 was found to promote the negative shift of PZFC [Figure 3A and B]. As for the higher electrocatalytic performance of Pt(111)/Ni(OH)2 than Pt(111), the modified Ni(OH)2 can lower the energy barrier of hydrogen adsorption and, thus, hasten the RDS kinetics of alkaline HER. Based on this, a model involving interfacial water was used to explain the different PZFCs. The recombination energy barrier of interface water is related to the strong interaction between water molecules and interface fields, which has a certain regulative effect on charge transfer within the electric double layer.
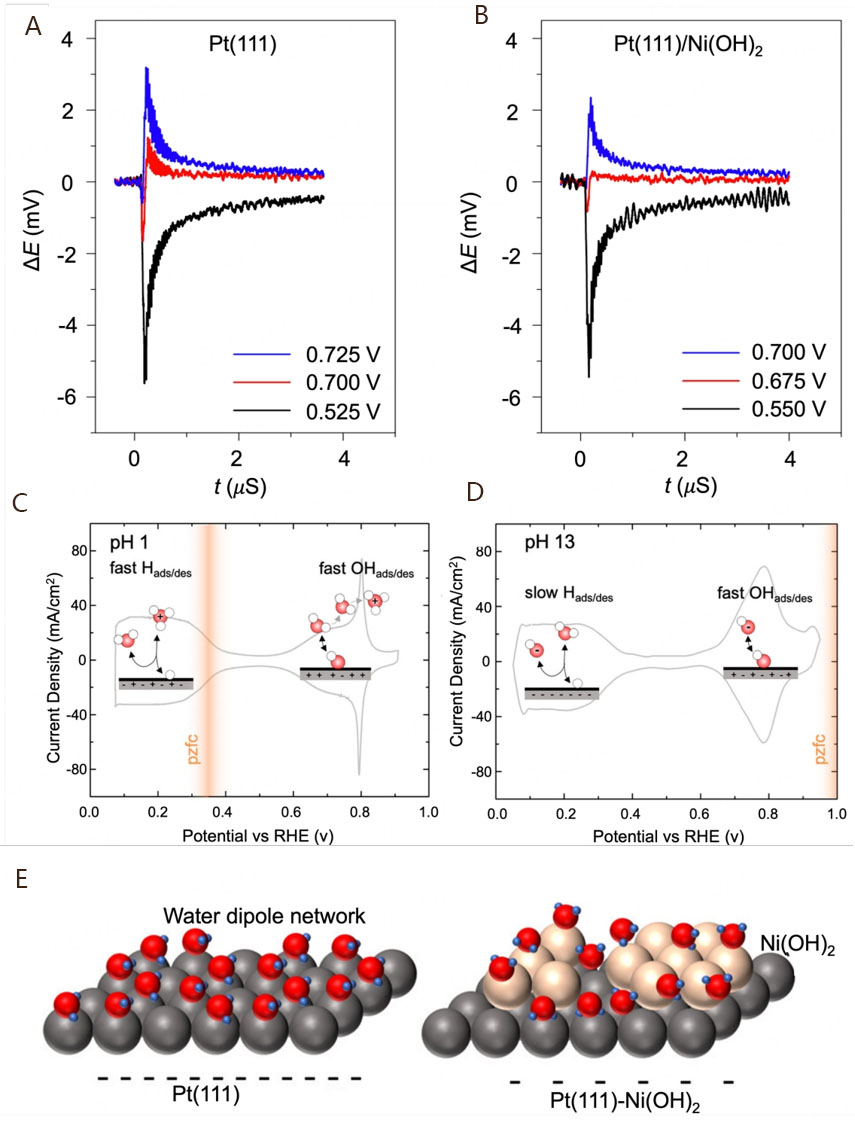
Figure 3. Laser-induced coulostatic potential transients collected for the Pt(111) electrode (A) and the Pt(111) electrode decorated with Ni(OH)2; (B). Reproduced with permission from[66]. Copyright 2017, Springer Nature; Cyclic voltammograms of Pt at pH 1 (C) and pH 13 (D) (the schematics show the different H and OH adsorption kinetics on the electrode surface). Reproduced with permission from[67]. Copyright 2020, American Chemical Society. (E) Schematic of the network of water dipoles on Pt(111) and the unstructured network on the Ni(OH)2 modified surface. Reproduced with permission from[68]. Copyright 2019, American Chemical Society.
Generally, PZFC is the inherent property of electrode materials. However, the discrepancy between hydrogen equilibrium potential and the PZFC of electrode materials during electrocatalytic reactions will strongly influence the electrocatalytic performance. According to the electrochemical phase diagram, the hydrogen equilibrium potential undergoes a shift with increasing pH, increasingly moving away from the PZFC [Figure 3C and D][67]. This discrepancy leads to an enhancement in the recombination energy of water. The enhanced energy barrier impels the formation of a strong network structure of water molecules in electrolytes, making it difficult for OH species to cross over to reach the interface electric double layer. This transfer process is adverse to the Heyrovsky/Volmer step, which results in sluggish alkaline HOR electrocatalysis. Therefore, the larger discrepancy between hydrogen equilibrium potential and PZFC in an alkaline medium than in an acidic medium may account for the differences in catalytic activity. As for the Pt(111) electrode, decorating Ni(OH)2 on its surface is beneficial for changing the PZFC to approach hydrogen equilibrium potential under alkaline conditions, which reduces the recombination energy barrier of water and thereby facilitates the charge transfer process of electrocatalytic kinetics.
In addition, the impact of the concentration of Ni(OH)2 on the alkaline HER performance was explored to further elucidate the PZFC theory associated with several other essential factors[68]. It was found that the PZFC exhibits a negative shift with increasing coverage of Ni(OH)2 on the surface of Pt(111) electrodes, thereby weakening the strength of the interface field. The weakened interface field strength is instrumental in the recombination of interface water structures [Figure 3E], thus favoring the charge and mass transfer process. Moreover, the weakened interface field strength also indicates the gradually positive net charge of the HUPD region. The positive net charge on the electrode surface helps to reduce the adsorption potentials for OH− and/or other anions, facilitating the RDS. Under alkaline conditions, the electrode surface is always negatively charged because of a much higher PZFC than the hydrogen equilibrium potential, suggesting that the specific adsorption of cations would affect the catalytical performance[69,70]. As a result, the PZFC is closely connected with several essential factors involving the electric double layer, water structure, interface electric field, ion effects, etc. In most cases, these theories need to be simultaneously used in alkaline electrocatalytic conditions, inducing the complexity and controversy of the reaction mechanism. Nevertheless, advanced experimental characterization techniques and theoretical calculations are effective methods for the exploration of mechanisms[71-74], which is conducive to guiding the design of excellent catalysts.
PGMs-BASED CATALYSTS DESIGN STRATEGIES
According to the different electrocatalytic reaction mechanisms above, HBE theory is a key factor for the rational design of HOR electrocatalysts. The indispensable Had intermediates demonstrate that the best ΔGH follows the Sabatier principle; i.e., the adsorption ability of Had intermediates should be moderate, neither too strong nor too weak[20,21]. As the ΔGH of PGMs is located near the apex of a volcano-like curve, PGM-based catalysts generally exhibit excellent HOR electrocatalytic performance. Meanwhile, in alkaline conditions, the participation of OH−, the variation of hydrogen equilibrium potential, the PZFC, and many other essential factors will all affect their alkaline HOR electrocatalytic reaction rates. Although still in dispute, these theoretical models have effectively guided the design of high-efficiency catalysts, such as the introduction of oxyphilic active sites, including Ni(OH)2 and Ru species, to improve the OHBE[55]. Given the low reserves and high prices of PGMs, feasible strategies must be taken to design high-performance and cost-effective catalysts for alkaline HOR under the guidance of theoretical mechanisms. Generally, regulating the electronic structures of catalysts has shown a remarkable relationship with their HBE and OHBE by changing the chemical and coordination environment of active sites, and increasing the number of these sites together helps to improve the total electrocatalytic performance[59,75-79]. The correlated design strategies include the facet/phase transition, the ligand/strain/ensemble effect, the size/morphology control, and so on. This section will provide a detailed description of the catalyst design strategies involving crystal structure engineering, size and atomic dispersion, low-dimensional, multicomponent atomic arrangement, surface active layer, and metal/support active interface construction.
Crystal structure engineering
Atomic arrangement for different facets
It is well noted that the electrocatalytic performance is strongly linked with the active sites of catalysts. In most cases, different atomic arrangements on the catalyst surface lead to distinct crystal faces and, thus, induce the exposure of diverse active sites[80]. This should be mainly due to the different coordination numbers of the outermost atoms on the crystal faces. For example, the face center cubic (fcc) structure exhibits three low-index facets of (111), (100), and (110), and the corresponding coordination numbers are 9, 8, and 7, respectively[30,81,82]. These distinct coordination numbers can lead to unique electronic and orbital configurations of the active sites by the variation of d-band density of states and bandwidth[31]. Extensive theoretical studies have demonstrated that the d-band center of surface active sites is directly associated with the binding energy of adsorbates. The different positions of d-band centers will lead to a change of binding energy strength, which will ultimately affect the adsorption strength of reactive sites on intermediates[83,84]. Due to the special adsorption ability of reaction intermediates on a well-defined crystal surface, the exposed crystal face may have a significant impact on the electrocatalytic performance[85-87]. For the alkaline HOR, exploring the electrochemical performances of Pt model catalysts with well-defined facets by experiment and theoretical calculations should be the prerequisite for designing excellent PGM-based catalysts. There are three most stable crystal facets of Pt, including Pt(111), Pt(100), and Pt(110) [Figure 4A]. Based on DFT calculations, Skúlason et al. compared the HER/HOR activities of Pt electrodes with different facets and found the most important activity descriptor should be the binding free energy of H[88]. Figure 4B shows that the energies of H adsorption are apparently different on the three most stable crystal facets. When H coverage is no more than 1 ML (1 H per 1 surface metal atom), the H adsorption energies of Pt(hkl) facets from negative to positive are in the sequence of Pt(100) < Pt(111) < Pt(110). However, the adsorption energy on Pt(111) facets becomes especially positive after exceeding 1 ML H coverage, indicating that the adsorption strength of intermediates is also related to their coverage on the catalyst surface. It is worth noting that the different energies of H adsorption on Pt(hkl) facets can lead to different HOR electrocatalytic performances, mainly attributed to the various geometric configurations of active sites on Pt(hkl) facets. Earlier, when taking the Pt single crystals as examples, Markovića et al. found that the surfaces of Pt(hkl) facets possess two different types of Had, including the HUPD at a high binding energy state and the overpotential deposited hydrogen (HOPD) at a low binding energy state[89]. These Had reaction intermediates exhibit different coordination structures on the surface of each Pt(hkl) facet, which accounts for the significant differences in alkaline HOR activity in the sequence of Pt(111) ≈ Pt(100) ≪ Pt(110) [Figure 4C]. When at higher potentials, the HOR activities of Pt(100) and Pt(110) significantly decrease due to the occurrence of OH adsorption (OHad). The inhibiting effect of OHad on the catalytic performance follows the sequence of Pt(100) ≪ Pt(110) < Pt(111). The results indicate that the well-defined Pt(hkl) facets exhibit different electrocatalytic performances, which can be attributed to the distinct states and coverages of HUPD and OHad under the corresponding potentials.
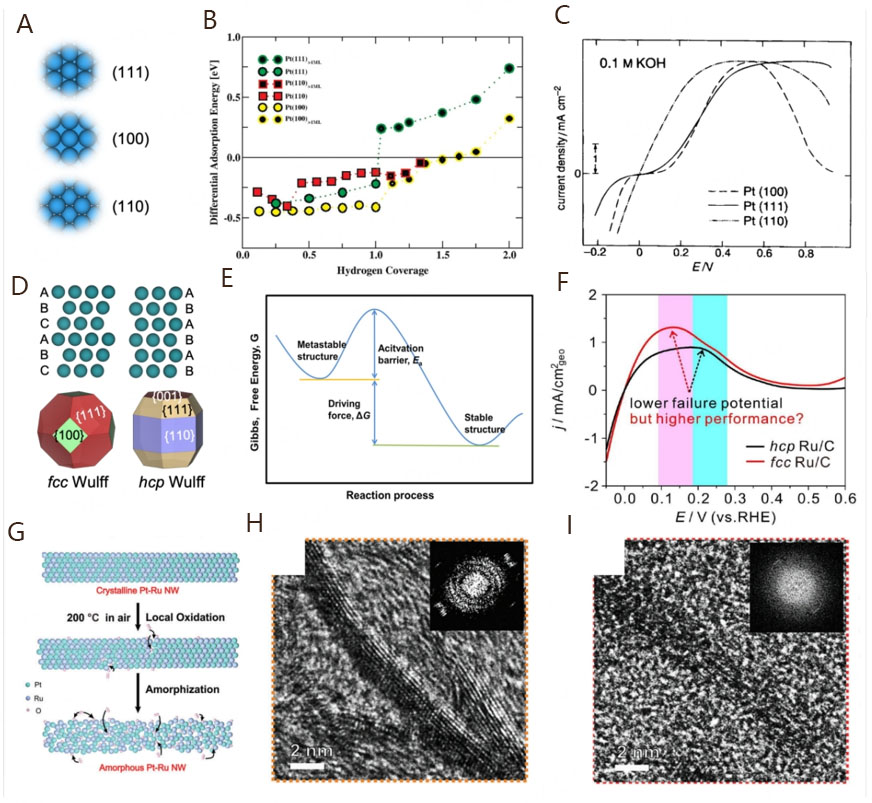
Figure 4. (A) Crystal models of Pt(111), Pt(100), and Pt(110) facets; (B) Differential energy of H adsorption as a function of H coverage for the Pt(111), Pt(110), and Pt(100) facets. Reproduced with permission from[88]. Copyright 2010, American Chemical Society; (C) Polarization curves for the HER/HOR on Pt(hkl) in 0.1 M KOH. Reproduced with permission from[89]. Copyright 1996, Royal Society of Chemistry; (D) Wulff polyhedral nanocrystal models with fcc and hcp phases; (E) Free-energy diagram over the transition from a metastable phase to a stable phase. Reproduced with permission from[31]. Copyright 2019, Elsevier; (F) HOR polarization curves of hcp and fcc- Ru/C. Reproduced with permission from[108]. Copyright 2021, Elsevier; (G) Cross-section schematic illustration showing the local oxidation-induced amorphization process; HRTEM images and corresponding fast Fourier transform (FFT) patterns (inset) of crystalline Pt53Ru47 NWs/C (H) and amorphous Pt53Ru47 NWs/C (I). Reproduced with permission from[117]. Copyright 2023, John Wiley and Sons.
Atomic stacking behaviors for different phases
To obtain more stable facets with different (hkl), the phase of a catalyst other than the fcc structure should be explored as well. It is well known that the phase of bulk metals depends on the stacking sequence, generally involving the fcc, hexagonal close-packed (hcp), and body-centered cubic packed (bcc) structures[90,91]. The space utilization rates of fcc and hcp are the highest at 74%, while the space utilization rate of bcc is 68%, intimating that most metal catalysts are prepared to deliver the most stable packed structures such as the fcc or hcp structure. According to the Wulff polyhedral nanocrystals [Figure 4D], the most exposed facets of the fcc structure are (111) and (100), and the most exposed facets of the hcp structure are (001), (111), and (110). The diversity of facets on different phase structures expands the space for improving the electrochemical performances of metal catalysts. Therefore, manipulating the planar stacking sequence of the target nanocrystals is another effective approach to designing excellent electrocatalysts[92,93]. However, most metal crystals tend to form a thermodynamically stable phase under normal conditions. It is difficult to obtain a novel crystal phase with abnormal stacking unless modifying the nucleation steps at atomic levels. Several recent studies have demonstrated that PGMs with an unusual crystal phase could be successfully synthesized by controlling the thermodynamic and/or kinetic processes during the structural transformation of nanocrystals[94-101]. Figure 4E shows the main factors for phase transition involving the thermodynamic driving force represented with ΔG and the kinetic activation energy barrier for atomic diffusion (Ea)[31]. The competition between ΔG and Ea determines the phase structure of nanocrystals to be stable or metastable. Thermodynamically, the stability of a crystal phase is related to the corresponding crystal facets that exhibit different surface energies. The crystal phase with the lowest Gibbs free energy should be the most stable structure, which can be determined by the volume and surface free energy[102,103]. To prepare a novel nanocrystal, the size should be the critical factor in determining the thermodynamically favorable phase transition in a certain growth circumstance, as the surface-to-volume ratio varies with the size of nanocrystals. For example, theoretical calculations have demonstrated that the thermodynamically stable phase for Rh nanocrystals is the fcc structure, while the one with hcp structures can be synthesized by decreasing the size to increase the surface-to-volume ratio[96,97]. This indicates that the thermodynamically favorable phase transition behavior can be achieved by modifying the nucleation and growth stage. However, from a kinetic perspective, it may not be easy to obtain a thermodynamically stable metal nanocrystal in some cases, as the growth is influenced by the concentration of metal atoms, which is linked with their diffusion/reduction/deposition rates[31]. The Ea for metal atomic diffusion is associated with atomic mobility, bonding energy, and the reaction temperature, and the reduction rate is determined by the reduction potentials. During the synthesis process of nanocrystals, corresponding conditions are usually controlled to reduce the atomic kinetic energy barrier and promote the formation of target products, such as improving the reaction temperature and using reductants[31,104,105]. As a result, products with various phase structures can be controllably synthesized based on changing thermodynamic and/or kinetic energy barriers.
In terms of electrocatalytic application, metal catalysts with the most thermodynamically stable phase structure may deliver unsatisfying performances due to the low proportion of exposed active sites on a perfect nanocrystal surface. In contrast, catalysts with metastable phase can expose abundant active sites due to the defective structure[106], which encourages researchers to carry out extensive exploration on the metastable phase structures. For example, the hcp structure of Ru nanocrystals is thermodynamically favorable under normal synthesis conditions. In 2013, Kusada et al. first discovered the pure metastable fcc structure of Ru NPs through a simple chemical reduction method and found the ones exhibit much higher catalytic performance in CO oxidation than hcp Ru counterparts[107]. This new discovery breaks the traditional beliefs that the anomalous phase structure can be achieved merely under extreme conditions. Concerning HOR electrocatalysis, Zhao et al. also synthesized fcc Ru and hcp Ru NPs with similar particle sizes by controlling the preparation process toward alkaline electrocatalysis[108]. Specifically, the fcc Ru/C with defects (twin crystal structure and/or stacking fault) exhibits easier oxidation of Had and higher activity than hcp Ru/C [Figure 4F]. This indicates that metal catalysts with metastable phase structures are effective candidates for alkaline HOR electrocatalysis due to the various structure defects[109].
As the performance associated with the exposed active sites, a unique phase regarding the amorphous structure is designed and has attracted increasing attention in recent years due to the abundant accessible polytype sites. In comparison with their crystalline counterparts, amorphous phases also exhibit many other advantages, including more flexible atomic-coordination ability and stronger corrosion resistance[110,111]. These merits endow amorphous metal materials with giant applicable potentials in electrocatalytic domains. However, the synthesis of amorphous metal materials is challenging. The forming ability is related to many factors, including electron concentration, atomic size, and the |ΔG| between liquid and solid states
Size and atomic dispersion
For nanomaterials with fixed crystal structures, regulating the particle size is not only beneficial for the adequate contact between surface atoms and the electrolyte and reactants but also improves the proportion of unsaturated coordination numbers such as the vertex or edge sites on the atomic surface. These merits can significantly improve the electrochemical active surface areas (ECSAs) and, thus, induce the nanomaterials with excellent specific and/or mass activities. Generally, particles with reduced size are reported to exhibit improved performance, while in some cases, it can decrease the specific activity of catalysts[118,119]. The much smaller NPs are susceptible to aggregate due to the much higher surface energy, indicating that particles with a high atomic dispersion are not easy to achieve. Nevertheless, nanoclusters and single atoms with much higher atomic dispersion could be successfully explored by engineering their coordination structures on functional supports. Moreover, for polycrystalline nanomaterials with various crystal facets, reducing their sizes may not improve the overall catalytic activities. Therefore, in this section, the size effect of aggregative NPs on the proportion of different facets and edge sites, the mass and/or specific activity, and the successful preparation of high-efficiency nanocluster and single-atom catalysts with a high atomic dispersion by regulating the coordination interactions with a given substrate will be discussed.
Size effect of aggregative nanoparticles
Since the electrochemical performance is closely related to the active sites on the catalytic surface, the nature and number of the reactive sites should be extensively explored. According to the crystal structure, the surface facet sites show an important influence on the electrocatalytic activity due to the different coordination numbers of the outermost atoms[30,81,82]. Relevant reports have demonstrated that the size effect is related to the binding energy of intermediates at different active sites such as the Pt sites on NPs[120]. For example, Shao et al. described the size effect of Pt truncated octahedral NPs on the dispersion and the number of surface sites in the range of 0.8-8 nm[121]. Specifically, increasing the particle sizes results in an obviously decreased site dispersion and number of edge sites on the surface, whereas the numbers of surface {111} and {100} sites are inversely increased with the particle sizes [Figure 5A]. Since these sites possess different coordination numbers, the various proportions of sites based on the size effect of Pt NPs will eventually affect the binding energy to reaction intermediates, thus influencing the electrocatalytic activities. It should be noted that the sites with lower coordination numbers would cause stronger binding energy to intermediates due to the upshifted d-band center[84]. However, the Pt NPs have been reported to show increased compressive strains as the particle size decreases, which suggests the corresponding weakened binding energy to intermediates[122]. Figure 5B shows the HOR mass activity and specific activity of Pt/C catalysts with different particle sizes. From 2.0 to 4.0 nm, the mass activities present a volcano-like curve with a maximum value of ~3.1 nm, while the specific activities increase with the particle sizes[23]. Similar changing tendencies of electrocatalytic activities against the Pt particle sizes have also been reported, where the specific activities will reach the plateau value if the particle sizes continue to increase[123]. However, the size effect for Pt NPs may not be applicable for other PGM NPs. Taking Ru NPs as examples, the specific activities of Ru/C also deliver a volcano-like curve with the increasing of Ru particle sizes, which is quite different from that of Pt NPs [Figure 5C][23]. Upon combination with the electron microscopy characterization, Ohyama et al. found that the increasing specific activity of Pt NPs should be attributed to the increased {111} and {100} sites on the surfaces that are more active for HOR performance than the surface edge sites[23]. For Ru NPs, the crystallinity and facets are not well-defined at the size of < 3 nm. The amorphous-like surfaces of Ru NPs give rise to an over-high fraction of unsaturated Ru atoms, which may be oxidized to reduce the specific activity. When the size is larger than 3 nm, the fraction of unsaturated Ru atoms on the well-defined facets becomes much lower, which is still adverse to the specific activity.
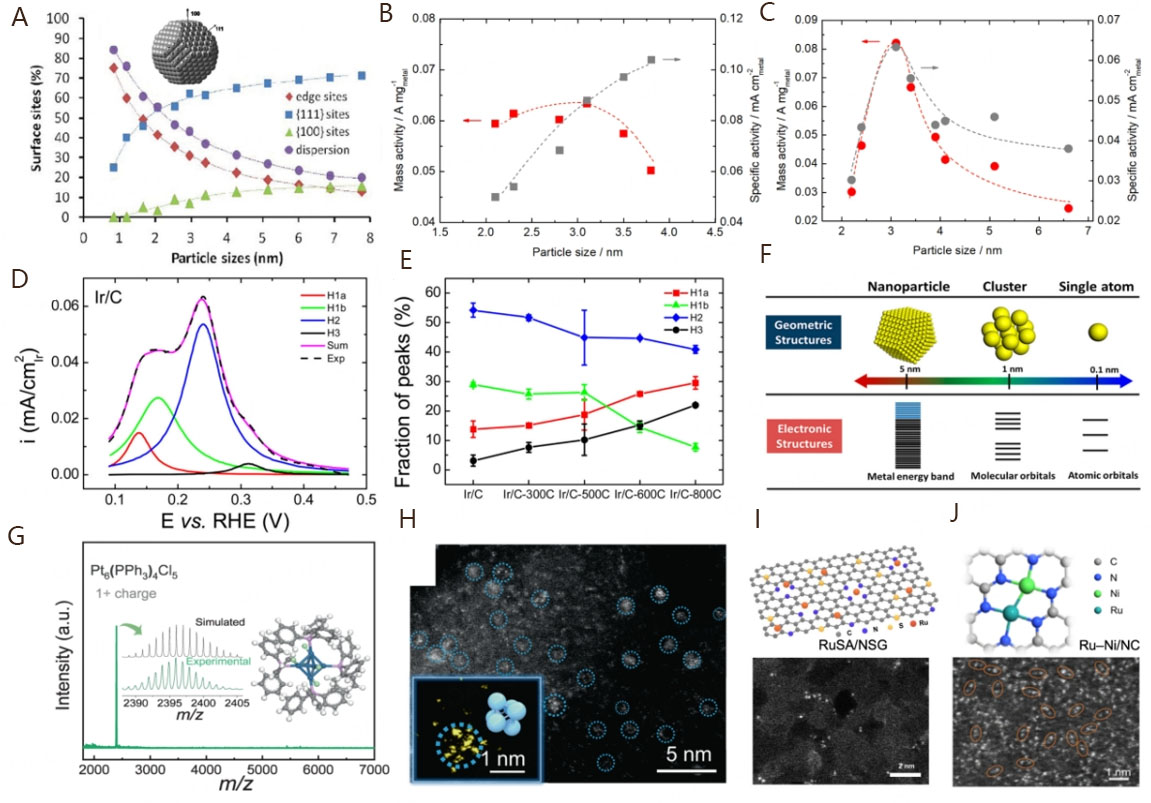
Figure 5. (A) Size dependence of dispersion and surface percentage of atoms on the {111}, {100} facets and the edges between the facets of a truncated octahedral Pt particle. Reproduced with permission from[121]. Copyright 2011, American Chemical Society. Plots of the mass and specific activities of the Pt/C (B) and the Ru/C (C) against the mean particle size. Reproduced with permission from[23]. Copyright 2013, American Chemical Society. (D) Deconvolution of hydrogen desorption peaks into four peaks centered at 0.13 V (H1a), 0.18 V (H1b), 0.24 V (H2), and 0.32 V (H3) for Ir/C; (E) Fraction of peaks H1a, H1b, H2 and H3 among Ir/C with different particle sizes. Reproduced with permission from[126]. Copyright 2015, American Chemical Society. (F) Geometric and electronic structures of single atoms, clusters, and nanoparticles. Reproduced with permission from[127]. Copyright 2018, American Chemical Society. (G) Electrospray ionization mass spectrometry of Pt6 nanoclusters (NCs); (H) Aberration-corrected HAADF-STEM image of Pt6 NCs/C; inset: the magnified image of a Pt6 NCs and corresponding model. Reproduced with permission from[137]. Copyright 2022, Springer Nature. (I) Atomic structure diagram and magnified HAADF-STEM image of the RuSA/NSG. Reproduced with permission from[142]. Copyright 2022, Springer Nature. (J) The optimized structure and atomic-resolution HAADF-STEM image of Ru-Ni/NC, in which some of Ru-Ni diatomic pairs are highlighted by orange ovals. Reproduced with permission from[143]. Copyright 2022, AAAS.
The size effects for Pt NPs and Ru NPs indicate that the alteration of particle sizes can regulate the type and number of surface active sites, including the terrace, edge, and corner sites[124,125]. Not all the active sites possess excellent electrocatalytic performance. For alkaline HOR electrocatalysis, it is necessary to systematically study how these active sites affect its sluggish reaction kinetics. Based on the reaction mechanism, HBE and OHBE theories are the key active descriptors to be applied to qualitatively analyze the activities of active sites, which can be presented by the electrochemical polarization curves. The active sites with various local environments usually exist in polycrystalline structures, making polycrystalline NPs to be rational research objects. Taking polycrystalline Ir as an example, Strmcnik et al. reported that its alkaline HOR kinetic can be determined based on the polarization curve[55]. To present a detailed kinetic study, Zheng et al. explored the HER/HOR polarization curves of Ir/C catalysts with particle sizes in the range of 3-12 nm and found the activity is directly related to the lowest HBE sites[126]. They deconvoluted the HUPD desorption peak involving the total number of surface sites into four different H-binding sites, i.e., H1a, H1b, H2, and H3 [Figure 5D]. The peak areas corresponding to these four H-binding sites vary with the particle sizes of Ir/C catalysts, indicating that varying particle sizes lead to different HBE of Ir/C [Figure 5E]. After normalizing the exchange current density to the surface area of H1a, H1b, H2, and H3 and comparing their relationships with the total ECSAs, this group finally concluded that the weak H-binding sites (H1a) contribute significantly to the activity, suggesting that increasing the population of low HBE sites (such as the low-index facets) rather than total active sites is an effective strategy for designing high-performance PGM-based nanocatalysts.
Atomically dispersed nanoclusters and single atoms
In earlier years, the exploration of particles mostly concentrated on a size larger than 1 nm. Based on the limited resolution of available characterization techniques at the time, the metal particles smaller than 1 nm cannot be visualized. Nevertheless, researchers have already inferred that the electronic structures of metal particles with sizes from larger than 1 nm to smaller than 1nm should undergo a strong transformation
For PGM nanomaterials, the design from nanoscale to atomic levels can expose abundant surface active sites, effectively boosting the atomic utilization of PGMs. However, the synthesis of PGM nanomaterials at atomic levels with definite atom numbers and satisfactory surface structure is still a long-term challenge, most likely attributed to the high reactivity of newly generated PGM atoms that leads to their uncontrollable growth. Nevertheless, extensive experimental efforts are dedicated to preparing eye-catching PGM catalysts at atomic levels. Under a mild reduction environment, by using triphenylphosphine (PPh3) molecules to protect Pt atoms well dispersed on the carbon support, Wang et al. successfully prepared Pt6 nanoclusters with precise size control of ~ 1 nm [Figure 5G and H][137]. It has been reported that the energy band structure of metal nanoclusters is discrete, which can make the d-band center shift with the number of atoms and, thus, enable the modification of the adsorption ability for intermediates during the electrochemical reaction[138]. During the alkaline HOR electrocatalysis, the Pt6 nanoclusters exhibit significantly higher activity and stability than the commercial Pt/C and Pt NPs. Mechanism studies demonstrate that the self-optimized ligand effect from the PPh3 ligand and the precisely controlled six-Pt-atom structure synergistically play decisive roles in regulating the d-band center, HBE, OHBE, and formation energy of H2O, which, thus, optimizes the alkaline HOR electrocatalytic performance of Pt6 nanoclusters.
It is well noted that the successful synthesis of structurally stable atomic-level PGM nanoclusters or single atoms depends on their adjacent ligands and/or the anchor sites. An excellent substrate plays a decisive role in stabilizing the atomically distributed structure[139-141]. With this perspective, Zhang et al. successfully prepared atomically dispersed Ru species by anchoring Ru3+ onto a pre-prepared three-dimensional (3D) N and S co-doped graphene (NSG) frameworks with rich in in-plane holes (RuSA/NSG) [Figure 5I][142]. The structural stability is attributed to the strong coordination of N and S dual heteroatoms on porous graphene. For alkaline HOR electrocatalysis, RuSA/NSG exhibits much higher performance than the commercial Ru/C. XAS technology and DFT calculations confirm that the excellent activity should be attributed to the accelerated reaction kinetics on the well-designed Ru–N4–S2 active center, specifically manifested as the weakened HBE, strengthened OHBE, and lowered activation energy barrier for water formation. Besides the isolated sites of Ru species for excellent alkaline HOR electrocatalysis, introducing other metal atoms to form atomically distributed diatomic sites may deliver exceptional performance as well. The DFT calculations have revealed that the ΔGH and ΔGOH are stronger at isolated Ru, Ir, and Rh sites but weaker at isolated Ni, Pd, and Pt sites[143]. In order to obtain the catalytic sites with the best ΔGH and ΔGOH, Han et al. designed to combine isolated Ru and Ni sites to build a novel active center, i.e., Ru–Ni diatomic sites anchored onto N-doped porous carbon substrates (Ru–Ni/NC) [Figure 5J][143]. The Ru–Ni/NC catalyst exhibits excellent electrocatalytic performance for alkaline HOR, superior to the single-site counterparts (Ru/NC and Ni/NC), and even outperforms the Pt/C catalyst. Due to the synergistic interplay between Ru and Ni atoms, the Ru–Ni diatomic sites of Ru–Ni/NC promote the dissociation of H2, the adsorption of OHad, and the formation of H2O, thereby significantly enhancing the alkaline HOR performance.
Based on the above analysis, we can conclude the factors for the high catalytic performance of nanoclusters and single atoms. By binding to adjacent ligands and/or heteroatoms, metal sites with a precise number of atoms can be stably anchored onto a given substrate. This induces the self-optimization of electronic structures, effectively modifying the HBE and OHBE in alkaline HOR kinetics. As for the PGM-based nanoclusters and single atoms, the maximum utilization of metal atoms can be achieved and, thus, promote the contribution to ultra-high electrocatalytic activity of PGM-based nanocatalysts with ultra-low noble metal loadings.
Low-dimensional controlling
The design of PGM-based nanomaterials with different dimensions is another effective method to improve the utilization ratio of precious metal atoms that have received extensive attention. Compared with NPs, one-dimensional (1D) nanocatalysts deliver good resistance to aggregation and/or Ostwald ripening due to the unique anisotropic constructions, making the catalytic active surface present excellent stability during the electrocatalysis[144-146]. In addition, the features of a high surface-to-volume ratio and fast electron/mass transfer rate further ensure substantial reactive sites on the surface, which endows 1D nanocatalysts with huge potential in alkaline HOR electrocatalysis[147,148]. For example, Yang et al. successfully prepared 1D worm-like Ir-oriented nanocrystalline assemblies (Ir ONAs) in a microemulsion [Figure 6A][25]. The electrocatalytic activity of Ir ONAs is much higher than that of Ir NP counterparts, indicating the broad perspectives of PGM-based catalysts with 1D nanostructures in alkaline HOR electrocatalysis. The interconnected nanostructure with a high aspect ratio endows the 1D ultrathin Ir ONAs with a high proportion of low-index facets that are supposed to deliver lower HBE than the edge and/or vertexes atoms, suggesting the accelerated HOR kinetics on ultrathin Ir ONAs. Moreover, the worm-like morphology formed by the self-assembly of small NPs can produce abundant profitable defect sites and lead to advantageous effects on the performance[149-151].
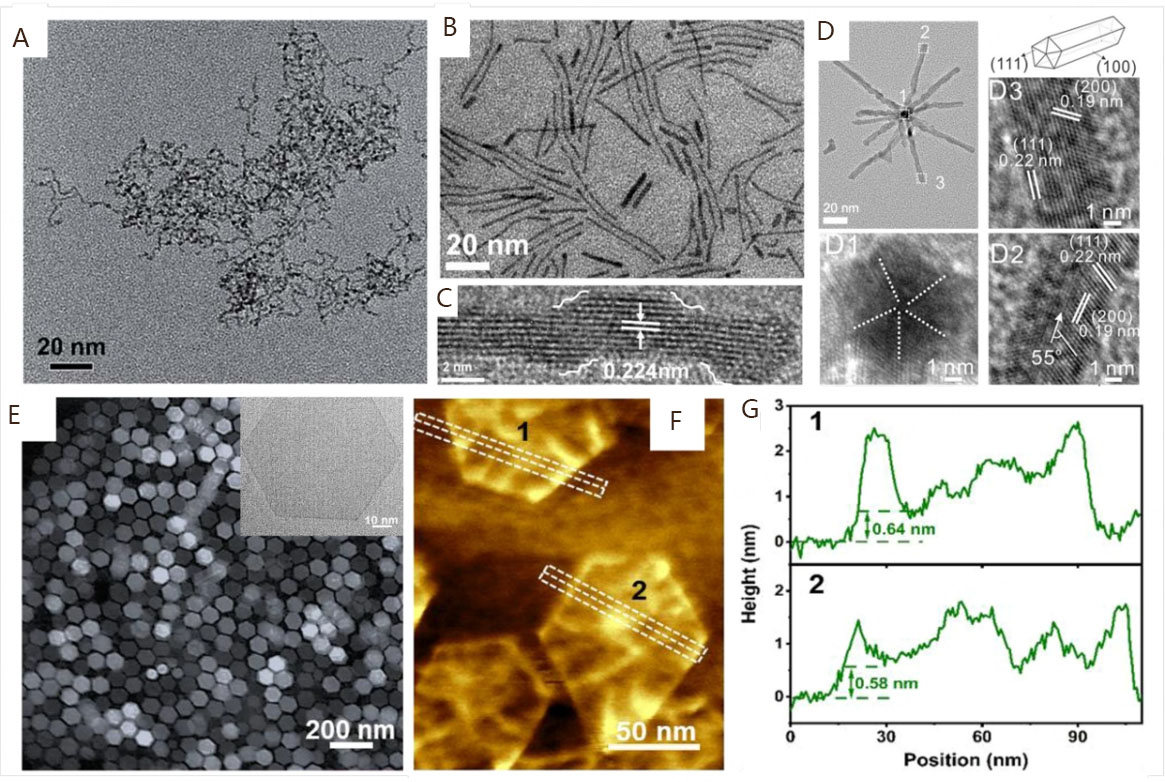
Figure 6. (A) TEM images of Ir-oriented nanocrystalline assemblies. Reproduced with permission from[25]. Copyright 2017, Royal Society of Chemistry. TEM (B) and HRTEM (C) images of Pt distorted nanowires. Reproduced with permission from[153]. Copyright 2022, Royal Society of Chemistry. (D) High magnification TEM image of cyclic penta-twinned Rh nanobranches and (D1-3) HRTEM images taken from different branches. Reproduced with permission from[154]. Copyright 2018, American Chemical Society. HAADF-STEM and TEM images (E), AFM image (F), and the corresponding height profiles (G) of RhMo nanosheets. Reproduced with permission from[164]. Copyright 2023, Springer Nature.
Since the self-assembling process of small NPs is susceptible to the reduction rate of metal precursors[152], an excessive rate may result in irregular growth behavior and, thus, influence the morphology of 1D worm-like nanostructures. Therefore, it is necessary to seek more effective synthetic strategies to prepare 1D nanostructures with more uniform morphology. Recently, Wang et al. successfully prepared Pt distorted nanowires (Pt DNWs) through a facile colloidal synthetic approach in oleylamine reaction solvents[153]. The Pt DNWs exhibit a uniform distribution with a morphology resembling lines, significantly different from the worm-like structure [Figure 6B]. As for the distorted NWs, there are abundant defects and edge step atoms on the surface, which is conducive to improving the atomic utilization [Figure 6C]. As expected, the alkaline HOR electrocatalytic activity of Pt DNWs/C is much higher than that of Pt/C, suggesting the superior advantage of the distorted NWs structure. It can be concluded that the high proportions of unique facets, profitable defects, and reactive atomic sites of 1D nanostructure provide a feasible platform for achieving the high atomic utilization of PGM-based nanocatalysts. By engineering the surface structure, Zhang et al. designed a facial solvothermal method to prepare Rh nanobranches (NBs) consisting of 1D ultrathin cyclic penta-twinned (CPT) nanorod subunits [Figure 6D][154]. The subunits grow along the <110> direction, and the sides and ends are bounded by {100} and {111} facets. The as-prepared Rh NBs possess a large percentage of well-defined {100} facets and twinned defects on the surface of CPT nanorods, which is considered beneficial for the exposure of numerous active sites. Additionally, the unusual intrinsic lattice strains of CPT nanorods also modify the surface electronic structure, laying the foundation for the superior electrochemical performances of 1D PGM-based nanocatalysts.
Besides 1D nanomaterials, ultrathin two-dimensional (2D) PGM nanomaterials with atomic thickness are also recognized as promising candidates with superb performance owing to their largest amplified ratio of exposed atoms, extended basal surfaces, and excellent conductivity[155,156]. However, the non-directional bonds of metallic materials make them more inclined to form a 3D close-packed structure, which leads to the challenging fabrication of 2D nanostructures. Nonetheless, a number of PGM nanomaterials with 2D superstructures have been successfully synthesized with unique electronic structures and potential electrochemical properties compared with their counterparts, including freestanding ultrathin NSs[157-160], mesoporous NSs[161], wrinkled NSs[162], metallene[97], and beyond. For instance, by using carbon monoxide as a surface confining agent, Huang et al. successfully prepared freestanding hexagonal Pd NSs less than ten atomic layers thick[157]. Their electrocatalytic activity toward formic acid oxidation is 2.5 times that of commercial Pd/C. Kong et al. reported the synthesis of ultrathin Ru NSs with thicknesses ranging from 1.0 to 1.2 nm, which exhibits much-enhanced performance toward water splitting compared with Ru powder counterparts[158]. The successful formation of Ru NSs is attributed to the self-decomposition and reduction of Ru acetylacetonate, the structure direction of isopropanol, and the structural assistant and stabilization of urea. To fabricate NSs with atomic thickness, Duan et al. used poly(vinylpyrrolidone) (PVP) as a ligand to stabilize metal atoms and successfully synthesized PVP-capped ultrathin Rh NSs with a thickness of less than 0.4 nm, which demonstrates superior catalytic activities to the commercial Rh/C[97]. For the electrochemical application of 2D PGM nanomaterials, the pure PGM catalysts toward alkaline HOR electrocatalysis are rarely reported. Several PGM-based alloy catalysts and heteroatom-modified PGM catalysts have been reported to exhibit superior HOR performance, such as the Ru2Ni multilayered NSs[163], atomic-thick metastable phase RhMo NSs[164], and sulfate-functionalized Ru NSs[165]. For instance, Zhang et al. reported the synthesis of freestanding RhMo NSs with a unique metastable hcp phase structure for alkaline HOR electrocatalysis [Figure 6E-G][164]. The atomic thickness of NSs is around 0.57 nm, suggesting a large amplified ratio of exposed active atoms. As expected, the HOR mass and specific activities of RhMo NSs/C are 21.09 and 7.04 times higher than those of commercial Pt/C. For low-dimensional nanostructures, the exploration of novel 2D ultrathin PGM-based nanocatalysts at the atomic level in alkaline HOR electrocatalysis is still far from meeting the practical application needs. Interestingly, the ultrathin nature of 2D allows catalysts to derive many different structures during subsequent growth processes, such as the Ir NSs-assembled 3D Ir superstructures and Pd-assembled super-NSs[156,166,167]. Due to the maximized intrinsic features of the assembled ultrathin 2D nanostructures, this kind of lamellar-assembled superstructure endows PGM nanocatalysts with superb electrocatalytic performance. Therefore, the controllable preparation of PGM-based nanomaterials with a large surface-to-volume ratio, a high proportion of surface defects, and an atomic level thickness could lay the material foundation for excellent alkaline HOR electrocatalysis.
Multicomponent atomic regulation engineering
Multicomponent atomic regulation has long been validated to effectively improve the activity of reactive sites for PGM-based catalysts, such as modifying PGMs with foreign metal atoms to form PGM-based alloy nanocatalysts. Alloys typically exhibit different electronic and structural properties from their pure metal counterparts, which endows them with higher selectivity, activity, and stability during the catalytic process[168-170]. This multicomponent atomic regulation strategy is beneficial for reducing the loading of precious metals while keeping a superior electrocatalytic activity. In this section, based on the intrinsic nature of precious metal atoms, we will focus on discussing their nanoalloys, including the disordered and ordered atomic arrangement, from the macroscopic control of metal atoms to the microscopic control of active sites.
Alloys with disordered atomic arrangement
PGM-based alloys with disordered atomic arrangement possess the advantages of a regulated electronic structure due to the strong ligand effect and/or strain effect. Owing to the different electronegativity and atomic radius between these metal atoms, the electron migration and lattice strain affect the charge density and d-band center on the surface of alloys, respectively, thereby influencing the strength of HBE and/or OHBE at the catalytic reaction interface[171,172]. Generally, due to the different HBE and OHBE on the metal atoms, adjusting the foreign elements in alloys is the prerequisite for improving the alkaline HOR performance of PGM-based alloy catalysts. Taking Pt catalysts as an example, Scofield et al. prepared some PtM alloy NWs (M = Fe, Co, Ru, Cu, Au) with the same composition to explore the influence of alloy effect on the electrocatalytic performance [Figure 7A-C][29]. The electronegativity difference between Pt and foreign atoms suggests the significantly different electronic binding energies between PtM alloy NWs and the pure Pt NWs, indicating that the surface reaction sites of PtRu, PtFe, PtCo, PtCu, and PtAu alloy NWs would deliver different adsorption abilities for reaction intermediates [Figure 7A and B]. Earlier, Kandoi et al. calculated the corresponding HBEs with site preference on different near-surface Pt-based alloys by theoretical calculations[173]. The theoretical strength of HBE follows the sequence of PtAu (-3.01 eV) > Pt (-2.72 eV) > PtFe (-2.65 eV) > PtCo (-2.62 eV) > PtRu (-2.42 eV) > PtCu (-2.32 eV). Accordingly, Scofield et al. concluded that their HOR catalytic activities are closely related to the theoretical HBE values[29]; only appropriate weakening of HBE can improve the HOR catalytic kinetics of Pt-based catalysts [Figure 7C]. This illustrates that adjusting the kind of foreign elements in Pt-based alloys can optimize the HBE by strong electronic interactions and, thus, improve catalytic activity. The adoption of different foreign elements is also applicable to form other PGM-based alloy catalysts with superior HOR performance to their single metal counterparts, such as the IrM (M = Fe, Ni, Co) alloy NPs and RuM (M = Fe, Ni, Co) alloy NPs[59,174].
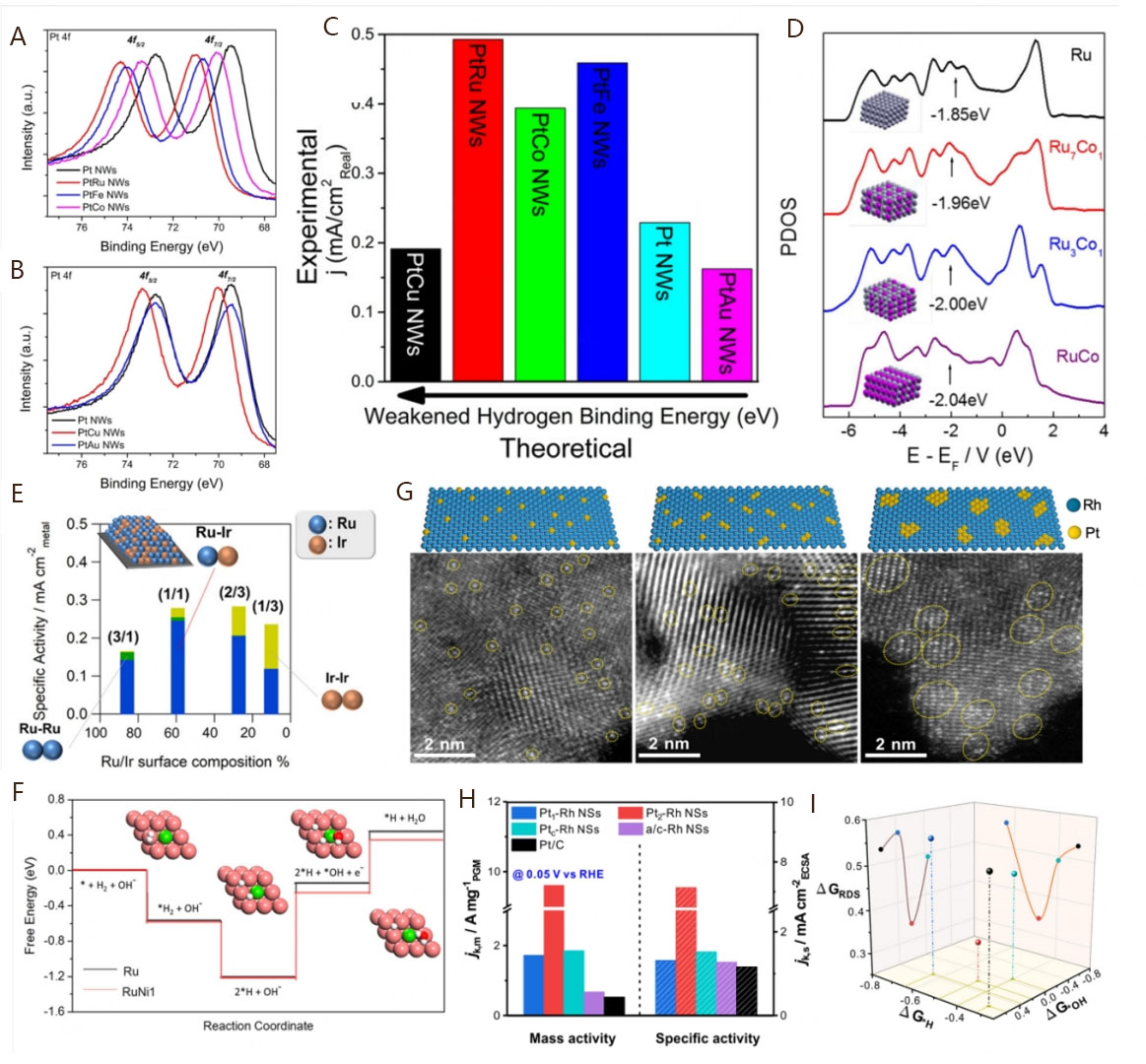
Figure 7. (A and B) XPS spectra of Pt 4f region for Pt NWs, PtRu NWs, PtFe NWs, PtCo NWs, PtCu NWs, and PtAu NWs; (C) Bar graph highlighting experimental HOR exchange current densities as a function of the calculated surface HBE values for models of “near-surface alloys”. Reproduced with permission from[29]. Copyright 2016, American Chemical Society. (D) Partial density of states of the Ru d-band for Ru, Ru7Co1, Ru3Co1, and RuCo. The Ru d-band centers are indicted in the figure. Reproduced with permission from[175]. Copyright 2020, American Chemical Society. (E) Relationship between the HOR-specific activity and the Ru/Ir surface composition, where the Ru-Ir, Ru-Ru, and Ir-Ir pairs are colored in blue, green, and yellow, respectively. Reproduced with permission from[177]. Copyright 2020, American Chemical Society. (F) Free energy diagrams of the elementary processes of HOR on Ru and RuNi1, including atomic configurations of each state on various metal surfaces. Color: orange, Ru; green, Ni; red, O; white, H. Reproduced with permission from[185]. Copyright 2020, American Chemical Society. (G) The optimized structure and aberration-corrected HAADF-STEM images of Pt1-Rh NSs, Pt2-Rh NSs, and Ptc-Rh NSs; (H) HOR mass activities and specific activities in 0.1 M KOH electrolyte; (I) Plot of RDS free energy (ΔGRDS) scaling with ΔGH and ΔGOH on Pt1-Rh(111) (blue), Pt2-Rh(111) (red), Ptc -Rh(111) (green), and Pt(111) (black) surfaces. Reproduced with permission from[186]. Copyright 2023, American Chemical Society.
Since the H adsorption abilities of Fe, Ni, and Co atoms are moderately weaker than that of most precious metal atoms, alloying PGM with them to weaken the HBE of PGM-based alloy has become a widely recognized strategy. Besides the different HBEs on diverse foreign atoms, the composition of PGM-based alloys can also affect the catalytic activity due to the regulated surface d-band center[175]. Wang et al. explored the effect of Co/Fe/Ni content on the alkaline HOR performance of RuM (M = Co, Fe, Ni) alloy catalysts and found that a small amount of Co/Fe/Ni can significantly enhance the activity[174]. As the d-band center is correlated to the HBE, the partial density of states of Ru d-orbitals for RuCo alloys with different Co contents were calculated by DFT [Figure 7D]. The results show that the d-band center of Ru gradually shifts to lower energies as the Co content in RuCo alloy increases, indicating the corresponding weakening of the HBE of RuCo alloy catalysts. Similar results were also obtained on RuFe and RuNi alloys with different Fe/Ni contents, respectively. Owing to the ligand and strain effects induced from different Co/Fe/Ni contents, the HBE on Ru active sites of RuM alloys can be moderately weakened to obtain high-efficiency alkaline HOR performance.
Based on the above conclusion, it can be seen that the electrocatalytic performance of PGM-based alloys is influenced by the foreign atoms and their contents. These factors regulate the HBE of catalytic active sites through strong electronic interactions. Thus, extensive explorations, including precisely regulating the kind, microscopic structure, and number of catalytic active sites on the surface of PGM-based alloys, must be dedicated to ultimately boost the alkaline HOR electrocatalytic performance[176]. The PGM atoms, such as Pt, Ir, and Ru, have been reported to serve as the active sites for their corresponding alloy catalysts[29,59,174]. If they are combined to form active pair sites and act together during the electrocatalytic process, such alloy catalysts may yield more satisfactory results. According to the discussion on the alkaline HOR mechanism, the HBE and OHBE theories seem to imply the key role of two kinds of active sites in simultaneously adsorbing H and OH species. So, it is necessary to reveal the effects of the category and composition of surface active sites on the alkaline HOR electrocatalytic kinetics. It has been confirmed that the atomically distributed diatomic sites for Had and OHad, such as Ru-Ni active centers of Ru-Ni/NC[143], are beneficial for the improvement of alkaline HOR electrocatalysis due to their synergistic interplay, as mentioned in Section 3.2.2. To construct active pair sites of alloys, Ishikawa et al. developed a series of carbon-supported Ru−Ir alloy catalysts (Ru−Ir/C) possessing different surface compositions of Ru/Ir, which all exhibit superior alkaline HOR activities compared with the Ru/C and Ir/C counterparts[177]. The specific activities of diverse Ru-Ir/C alloy catalysts were found to have no collection with HBE but show a volcano-like relationship with Ru/Ir surface composition, intimating the distinct effects of surface Ru and Ir atoms on reaction kinetics. Note that the much higher oxophilicity of the Ru site promotes the adsorption of OH species that react with the H species adsorbed on neighboring Ir site, leading to the superior reactivity of Ru-Ir pairs than Ru-Ru and Ir-Ir pairs [Figure 7E]. Therefore, constructing abundance dual-active sites on the catalytic surface that can adsorb H and OH species separately is an effective strategy for achieving high-efficiency HOR performance. Considerable similar progress has been made in enhancing the HOR kinetics by alloying PGMs with a more oxophilic metal, such as IrMo[178], RuNi[163], and several different PtM (M = Ru, Mo, Ni, Fe, Co) alloys[29,55,76,179-181], validating the effective universality of dual-active sites.
In order to optimize the electrocatalytic performance, the PGM-based alloys with dual-active sites mentioned above typically control the composition of surface sites by adjusting the total elemental composition. However, this will result in the insufficient utilization of active sites and the inability to obtain specific microstructure of active sites, which, thus, affects the insight into improving catalytic performance at the molecular level. Meanwhile, the design of catalysts at the atomic level has been recognized to ultimately maximize the utilization of active sites, providing a powerful approach to prepare PGM-based alloys with isolated metal atoms that can be named single-atom alloy catalysts[182-184]. On one side, the isolated metal atoms can modify the electronic structure of adjacent host metal substrates and, thus, influence the binding energies for reaction intermediates. On the other side, the specific microscopic structure of single-atom alloy catalysts can be precisely tuned to gain insight into the relationship between the structure and electrocatalytic activity. For example, Mao et al. reported the preparation of isolated Ni atoms dispersed on Ru NSs (RuNi1 NSs) for excellent alkaline HOR electrocatalysis, with pure Ru NSs and RuNi bimetallic NSs as comparisons[185]. The electrochemical results show that the mass activity of RuNi1 NSs is 6.3 and 16.6 times higher than that of RuNi NSs and Ru NSs, respectively, indicating the unique structural advantages of the single-atom alloy catalysts. The XAS measurements validate that Ni atoms are highly dispersed on Ru substrate in the form of single atoms by a strong Ni-Ru interaction. DFT calculations reveal that the strong Ni-Ru interaction weakens the HBE and strengthens the OHBE on the surface of RuNi1 models, which promotes the subsequent water formation and desorption during the alkaline HOR mechanism [Figure 7F]. This result suggests that rationally designing single-atom alloy catalysts with a strong alloy interaction is beneficial for accelerating the RDS (Volmer step) of alkaline HOR processes.
Additionally, the alloy interaction between guest atoms and the host metal substrate can be optimized by precisely tuning the aggregation state of guest atoms. To obtain atomic-dispersed PGM-based alloys with different aggregation states of guest atoms, our group recently synthesized host−guest-type Pt-on-Ru alloy NSs with atomic-dispersed single-Pt atoms, dual-Pt atoms, and clustered-Pt atoms (denoted as Pt1-Rh NSs, Pt2-Rh NSs, and Ptc-Rh NSs, respectively) by simply using the ultrathin Rh NSs to capture different numbers of guest Pt atoms [Figure 7G][186]. These atomic-dispersed Pt-Rh NSs all exhibit much higher activities than pure Rh NSs and commercial Pt/C catalysts, with Pt2-Rh NSs delivering the highest activity [Figure 7H]. The different activities should be attributed to the different ΔGH and ΔGOH of Pt1−Rh NSs, Pt2−Rh NSs, and Ptc−Rh NSs [Figure 7I], indicating that both electronic structure and oxophilicity play indispensable roles in improving the electrocatalytic kinetics. Notably, the moderately weakened HBE and strengthened OHBE are conducive to promoting water formation and desorption, which, thus, endows Pt2-Rh NSs with the highest electrocatalytic activity for alkaline HOR processes. Therefore, precisely adjusting the aggregation states of guest metal atoms can effectively regulate the electronic effect on host metal atoms, coupled with the modified oxophilic effect, which, thus, jointly optimizes the HBE and OHBE toward superior alkaline HOR electrocatalysis. As a result, the alkaline HOR performance of PGM-based alloy catalysts can be improved from two aspects: (1) optimizing the electronic structures or d-band centers of active sites by regulating the type and composition of foreign metal atoms; (2) designing dual-active sites that can adsorb Had and OHad intermediates separately. This involves precise regulation of their microstructures and compositions at the atomic level to optimize electronic and oxophilic effects.
Intermetallics with ordered atomic arrangement
PGM-based alloys with ordered atomic arrangement are the ones with periodically arranged metal atoms under strict stoichiometry, which are generally defined as intermetallics [Figure 8A]. In comparison with the disordered atomic arrangement, intermetallics present significantly different physicochemical properties, even if they both possess the same elemental category and composition[187]. Intermetallics with a well-defined atomic arrangement can provide predictable electronic effects and uniform active sites on the surface, which is conducive to the precise regulation of structure and, thus, to the optimization of adsorption and activation ability for reactants. Additionally, the lattice mismatch in intermetallics can induce strengthened strain effects, leading to the modification of electronic structures[188]. Moreover, the well-defined strong atomic binding environment of intermetallics is especially beneficial for the compositive and structural stability, implying the broad potential in electrochemical applications[189]. Despite these advantages, the successful synthesis of intermetallics is still subject to limited experimental methods. Even though PGM-based alloys with ordered atom structures are thermodynamically favorable in some cases, the prevalent products synthesized by conventional chemical approaches are the ones with disordered atom arrangements[190,191]. Yan et al. summarized the thermodynamic-equilibrium phase diagram regarding the composition of ordered and disordered phases in A-B binary alloys against the temperature [Figure 8B][191]. The colored regions represent the thermodynamically-equilibrium phases, and the ordered intermetallics seem to be more favorable than the disordered solid solution. By controlling temperature, the ordered and disordered phase structures with the same atomic ratio may undergo mutual transformation. Furthermore, they plotted the Gibbs free energy (G) as a function of composition at two different temperatures [Figure 8C]. The thermodynamic stability is determined based on the variations in entropy (S) and enthalpy (H) with temperatures (ΔG = ΔH - TΔS). The disordered solid solution is more stable than the ordered intermetallics at high temperatures (> T1) due to the higher S of the disordered ones. At low temperatures

Figure 8. (A) The structure comparison of random alloy and intermetallics; (B and C) Schematic illustration of thermodynamic-equilibrium conditions for the formation of an intermetallic phase (AmBn) in the A-B binary system. Reproduced with permission from[191]. Copyright 2017, John Wiley and Sons; (D) HOR polarization curves of Ru3Sn7/C, RuSn/C, Pt/C, and Ru/C in 0.1 M KOH; (E) The density of states of Ru-d band and Sn-p band in Ru3Sn7 and RuSn; (F) The calculated ΔGOH of Ru, Ru3Sn7, and RuSn. Reproduced with permission from[206]. Copyright 2023, John Wiley and Sons.
For a real material system, the successful synthesis of a stable intermetallic compound is much more complicated, which is not only related to metal species, composition, and temperature but also matters with the particle size, morphology, and exposed facets[192-197]. Alloyeau et al. reported that the disorder/order phase-transition temperature of Pt-Co alloys decreases with the decrease in particle size due to a higher surface energy of small particles than the bulk[192]. As catalysts with different facets also exhibit distinct surface energies, the phase-transition temperature varies with exposed facets as well. With respect to the specific synthesis methods for the formation of intermetallics, direct annealing at high temperatures is one of the most effective approaches. Sun et al. synthesized the ordered PtFe nanocrystals by annealing the disordered fcc-PtFe nanocrystals at 600-700 °C[198]. Nevertheless, the high-temperature calcination can easily cause aggregation and sintering, which motivates the construction of supports and shell layers to protect intermetallics. For example, Wang et al. reported the synthesis of Pt3Co intermetallic NPs by a thermal treatment of Pt NPs supported on Co-doped metal-organic-framework-derived carbon at 900 °C[199].
For alkaline HOR electrocatalysis, extensive and systematic experimental research should be conducted to explore feasible methods for preparing PGM-based intermetallics with excellent electrocatalytic performance. Cable et al. reported the synthesis of intermetallic PtSn and PdSn NPs via a polyol process proposed by Schaak[204]. Owing to the modified electronic structure of Pt sites by the electron donation of Sn, the adsorption ability of intermetallic PtSn/C for Had can be optimized, thus leading to its superior HOR activity to the commercial Pt/C[205]. More recently, Su et al. prepared an ordered Ru3Sn7/C intermetallic compound through a simple colloid method and compared it with the disordered RuSn/C counterpart. The electrochemical experiment shows that ordered Ru3Sn7/C exhibits much higher catalytic activity for alkaline HOR than the disordered RuSn/C [Figure 8D][206]. Because of a well-defined atom binding environment, the ordered Ru3Sn7 alloy delivers a more intense d–p orbital hybridization than the disordered RuSn alloy, indicating a much stronger electron interaction between Ru and Sn in the ordered Ru3Sn7 [Figure 8E]. DFT calculations further revealed that the OHBE of ordered Ru3Sn7 is much stronger than that of disordered RuSn, which should be attributed to the ordered and periodic atomic arrangement that regulates the surface oxophilic ability for OH species [Figure 8F]. Thus, the excellent alkaline HOR electrocatalytic performance of Ru3Sn7/C intermetallic compounds can be attributed to the strong electronic and oxophilic effects induced by the specially ordered atomic structure. These advanced reports indicate that improving synthesis strategies to precisely regulate the atomic ratio, atomic arrangement, and exposed facets of PGM-based alloys to prepare well-defined intermetallics is particularly beneficial for efficient alkaline HOR electrocatalytic processes.
Metal-nonmetal compounds
Different from conventional alloys, some metal-nonmetal compounds also exhibit metallic properties. The nonmetal elements with large electronegativity induce partial electron localization, leading to the redistributed surface charges and, hence, the attractive catalytic behaviors, which have been widely used for HER. Among them, metal phosphides usually exhibit superior activity compared to their pure metal counterparts. However, the studies about the metal phosphides for alkaline HOR have been less reported until recently. A series of PGM (Ru, Rh, or Pd)-based phosphides towards hydrogen electrooxidation in alkaline media have been developed[60,207,208]. Typically, Typically, Zhao et al. reported that Ru2P exhibited about three times enhanced intrinsic activity compared to element Ru and was comparative to commercial Pt/C[207]. By using it as the anode, the AEMFC single cell showed a very high peak power density (PPD) value of 1.3 W cm-2. Similarly, Rh2P NPs reported by Yang et al. also exhibited significantly improved activity compared to pure Rh NPs[60]. Huang et al. adopted a defect-derived strategy for creating phosphorous vacancies (P-vacancies) on Rh2P to break the atomically homogeneous surfaces that are terminated by P atoms[209]. In this case, the sub-surface Rh atoms could be exposed to act as the exclusive sites for Had adsorption, decoupling the competitive adsorption of both Had and OHad on the same type of active centers. Therefore, the defective Rh2P nanowires exhibit extremely high activity for alkaline HOR, attaining 5.7 mass activity that of the Rh2P nanocubes with relatively perfect surfaces. In consideration of the polymorphic feature, palladium phosphides, including Pd3P, Pd5P2, and PdP2, have been successfully synthesized, in which the mass/specific activity follows the order of
Surface active layer
It is well known that the electrochemical performance of catalysts is directly related to the physiochemical properties of their surface atoms that are influenced by the internal atom structure. Designing multicomponent PGM-based catalysts with engineered surface active layers, such as the core-shell nanostructures, is an effective strategy to improve the surface electronic properties by the strain and ligand effects [Figure 9A]. The strain effect, including tensile strain and compressive strain, stems from the lattice mismatch between core and shell atoms that have different crystal structures or lattice parameters[211-213]. Generally, the strain effect plays a key role in regulating the position of the d-band, which is related to the adsorption ability of reaction intermediates. For PGMs with more than half-filled d-bands, the tensile strain can cause a narrowed bandwidth. While keeping the filling of the band fixed, narrowing the bandwidth would result in the d-band center shifting up toward the Fermi level, thereby inducing strong binding energy for reaction intermediates [Figure 9B and C][214]. With respect to the compressive strain, it generally results in a downward shift of the d-band center, which presents the inverse effect. In this regard, several studies have validated that tuning the strain on the active shell layer can improve the electrochemical intrinsic activity by modifying the electronic structures[212]. For example, Gong et al. explored the strain effects on Pt shells by controllable self-passivating of ordered intermetallic PtSb NPs (PtSb and PtSb2) under potential cycling to form core@shell Pt−Sb@Pt[215]. The successfully formed core-shell PtSb@Pt and PtSb2@Pt possess the Pt shells with a thickness of approximately 5-6 atomic layers, respectively [Figure 9D and E]. Alkaline HOR electrocatalysis indicates that the exchange current density of PtSb@Pt is 1.6 and two times larger than that of commercial Pt/C and PtSb2@Pt, respectively [Figure 9F]. Owing to the different crystal structures of PtSb and PtSb2 cores, the strained Pt shells of PtSb@Pt and PtSb2@Pt deliver 10% compressive and 4% tensile strain, respectively, leading to the opposite shift of d-band centers. The compressive strain endows PtSb@Pt with a downshifted d-band center and, thus, causes a weakened HBE on its surface, which is favorable for facilitating the HOR kinetics. In contrast, the tensile strain leads to a strengthened HBE on the surface of PtSb2@Pt, which is unfavorable for HOR.
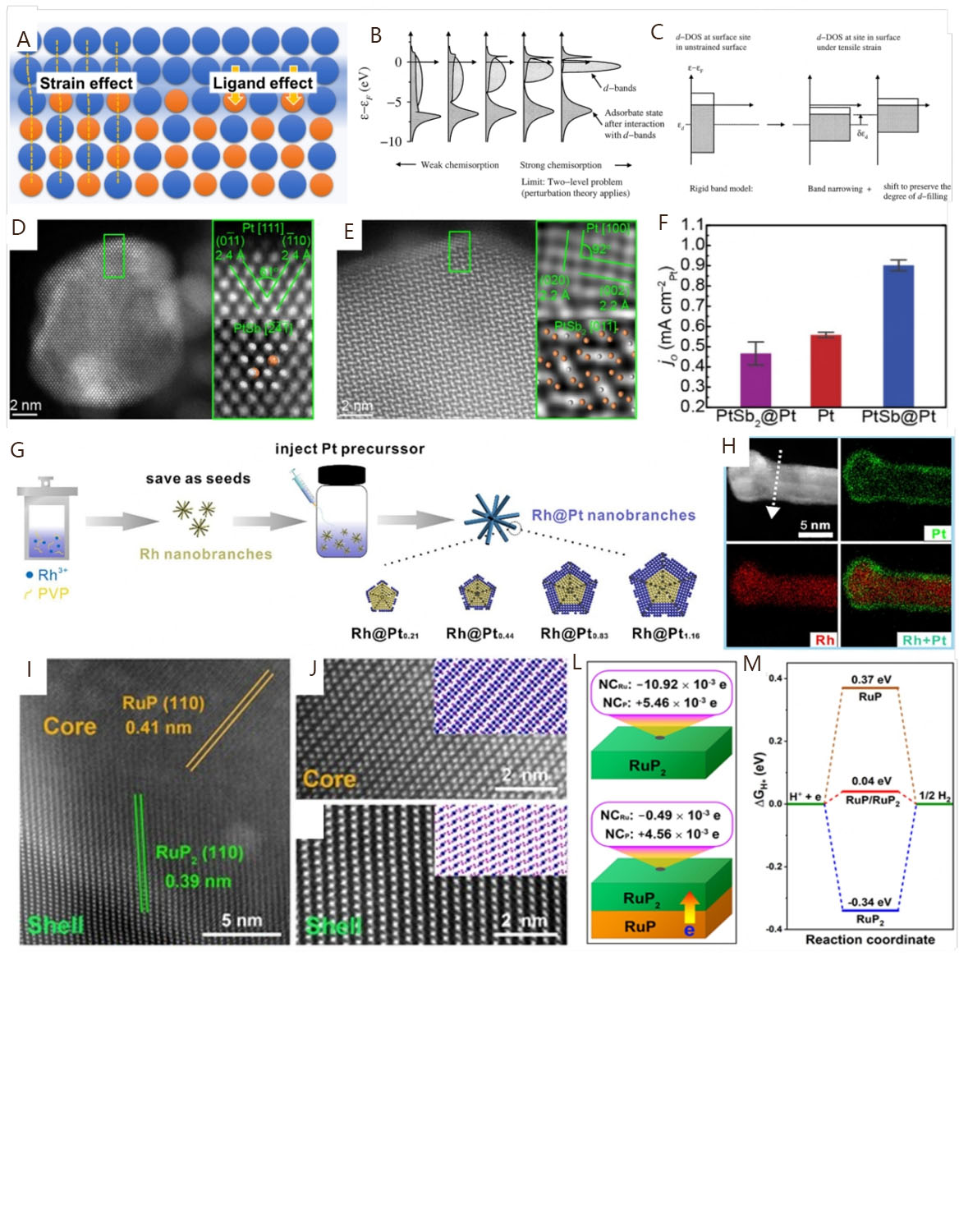
Figure 9. (A). Schematic illustration of strain and ligand effect in a core-shell nanostructure; (B) The local density of states projected onto an adsorbate state interacting with the d bands at a surface; (C) Illustration of the effect of tensile strain on the d band center. Reproduced with permission from[214]. Copyright 2000, Elsevier. (D) STEM/HRTEM image for PtSb nanoparticles after cycling, showing that a PtSb@Pt structure has formed; (E) STEM/HRTEM image for PtSb2 nanoparticles after cycling, showing that a PtSb2@Pt structure has formed; (F) the HOR exchange current densities of Pt, PtSb@Pt, and PtSb2@Pt. Reproduced with permission from[215]. Copyright 2023, American Chemical Society. (G) Schematic illustration of the preparation of Rh@Ptx NBs; (H) STEM and corresponding EDS elemental mapping of Rh@Pt0.83 NBs. Reproduced with permission from[217]. Copyright 2022, Elsevier. High-resolution ADF-STEM image of RuP@RuP2/C (I) and the atomic-resolution ADF-STEM image of RuP core (J) and RuP2 shell (K); (L) Net charge analyses of RuP/RuP2 and RuP2; (M) Calculated ΔGH of RuP/RuP2, RuP2, and RuP. Reproduced with permission from[218]. Copyright 2022, John Wiley and Sons.
Meanwhile, the strain effect on the active shell layer can also be optimized by controlling the thickness of the shell. Schwämmlein et al. prepared Ru@Pt core-shell NPs with different thicknesses of Pt shell and found that the alkaline HOR performances exhibit a volcano-like relationship with the Pt shell thickness from the sub-monolayer to the multi-layer[216]. Recently, our group designed a simple seed-mediated method to prepare penta-twinned (PTw) Rh@Pt core-shell NBs with a tunable Pt-shell thickness for alkaline HER/HOR electrocatalysis[217]. The Rh NBs were first synthesized to serve as seeds for the subsequent deposition of Pt shells, and the energy dispersive X-ray spectroscopy (EDS) elemental mapping images demonstrate the successful formation of Rh@Pt NBs with Rh-core and Pt-shell nanostructures
Besides the strain effect, the inner-interfacial interaction at core/shell interfaces may also play a key role in promoting the strong electron transfer of the surface active layer to influence its adsorption ability of reaction intermediates. Du et al. designed a homologous core-shell RuP@RuP2/C nanocatalyst that comprises a Ru-rich RuP core and a P-rich RuP2 shell layer [Figure 9I-K][218]. By utilizing melamine polyphosphate to release active phosphorus species slowly and persistently, RuP@RuP2/C can be successfully formed via a phase transformation mechanism. At a moderate concentration of Ru3+, the self-growth of RuP2 units and their transfer to form RuP achieve a good balance, thus forming a core-shelled RuP@RuP2 architecture. DFT calculations were performed to study the inner-interfacial interaction of RuP/RuP2, and the results show that the average net charge per atom of Ru (NCRu) and P (NCP) in RuP2 is significantly influenced by the incorporated RuP, suggesting that the electrons transfer from RuP to RuP2 [Figure 9L]. The redistribution of electron density in RuP/RuP2 reveals the strong electronic interaction between RuP cores and RuP2 shells, which is conducive to optimizing ΔGH to be nearly thermoneutral toward fast hydrogen adsorption /desorption kinetics [Figure 9M]. For electrochemical catalysis, RuP@RuP2/C exhibits the largest exchange current density in alkaline HOR electrocatalysis when compared to the RuP2/C, RuP/C, and Pt/C; Especially, its mass current density is even over two times higher than that of Pt/C. The excellent alkaline HOR performance of RuP@RuP2/C is attributed to the strong electron coupling at the interface of RuP/RuP2, corroborating the superiority of a core-shell architecture with an inner interface.
Metal/support active interface
In addition to the internal core/shell interface, an interfacial interaction typically comes from the external metal/support interface of heterostructures, such as the various interfaces between metal and oxides, hydroxides, carbon nitrides, etc[219-221]. The metal-support interaction has been investigated for many chemical reactions, such as O2 oxidation/reduction, CO oxidation, and NO reduction. For alkaline HOR electrocatalysis, the metal NP-(hydr)oxide support catalysts with a strong interfacial interaction play a crucial role in promoting the reaction kinetics since the metal/metal (hydr)oxide heterostructures typically possess dual-active sites on the surface for adsorbing Had and OHad intermediates. For example, Strmcnik et al. reported the preparation of Ni(OH)2/Pt(111) catalysts toward high-performance alkaline HOR electrocatalysis, with Pt atoms and Ni(OH)2 clusters serving as active sites for binding Had and OHad, respectively[55]. Kundu et al. reported Rh–Rh2O3 NPs/C to exhibit much higher HOR performance than the Rh2O3-free Rh NPs/C due to the synergistic interactions of Rh metal and Rh2O3[222]. Miller et al. reported the synthesis of Pd/C-CeO2 catalysts for high-performance alkaline HOR electrocatalysis[223]. They found that the HBE on the surface of Pd supported on C-CeO2 is weaker than that of Pd supported on pure C, indicating that the strong interactions between Pd and C-CeO2 support are conducive to the modification of surface electronic structures. More recently, this group synthesized a new type of CeOx-Pd/C catalysts by selective deposition of CeOx onto Pd/C NPs[224]. The CeOx-Pd/C delivers an ultra-high homogeneity of CeOx distribution and a high interfacial contact area between Pd and CeOx. The interfacial contact area can be quantitatively estimated to investigate its relationship with electrochemical performance. Interestingly, the calculated HOR-specific exchange current of the CeOx-Pd/C increases with the increase of interfacial contact area between Pd and CeOx, whereas the excess CeOx would depose onto the carbon to form large nanoislands that are detrimental to the activity [Figure 10A-C].
Figure 10. (A) High-resolution STEM maps of Pd and Ce of 0.24 CeOx-Pd/C, 0.38 CeOx-Pd/C, and 0.59 CeOx-Pd/C; (B) HOR polarization curves in 0.1 M KOH; (C) Interfacial contact area as a function of the Ce/Pd bulk atomic ratio. Reproduced with permission from[224]. Copyright 2020, John Wiley and Sons; (D) HRTEM images of Ru@TiO2; (E) Enlarged view of the interface between Ru and TiO2; (F) HOR polarization curves of Ru@TiO2, PtRu/Ccom, and Ru/C catalysts in 0.1 M KOH. Reproduced with permission from[226]. Copyright 2020, Springer Nature; (G and H) HAADF-STEM images of IrWOx/C, with amorphous WOx clusters (2,4) decorated on the Ir-W nanocrystallines (1,3); (I) Histograms of HOR mass activities and exchange current densities of IrWOx/C, Ir/C, and Pt/C. Reproduced with permission from[233]. Copyright 2020, Elsevier.
In most cases, interfaces formed by a traditional deposition method may exhibit weak interactions, which can easily result in poor structural stability[225]. Thus, novel synthetic strategies should be designed to construct metal/metal (hydr)oxide interfaces with strong interactions toward high-efficiency alkaline HOR. Zhou et al. reported the preparation of Ru@TiO2 with strong Ru–Ti bonds at the Ru/TiO2 interfaces by a novel lattice-confined strategy[226]. During the synthesis, amorphous urchin-like TiO2 with abundant defects was first prepared and then saturated with Ru3+. After H2 annealing at 500 °C, many Ru clusters were formed and anchored into the lattice of highly crystallized anatase TiO2. The Ru clusters were found to epitaxially grow along the lattice of TiO2 rather than physically adsorb or support on its surface, which generates a new crystal structure at the Ru/TiO2 interface to form abundant Ru–Ti bonds [Figure 10D and E]. Owing to the special lattice confinement at the interface, Ru clusters present strong electronic interactions with TiO2 substrates by the electron penetration from electron-rich TiO2 to Ru, which not only optimizes the binding energy for reaction intermediates but also enhances the antioxidation ability of Ru. During the alkaline HOR electrocatalysis, the Ru@TiO2 catalyst exhibits similar catalytic behavior to that of commercial PtRu/C, whereas the Ru/C catalyst presents rapid degradation in activity due to the irreversible oxidation of surface Ru active sites [Figure 10F]. The mass activity of Ru@TiO2 catalysts is 15.6% and 34.0% higher than that of PtRu/C and Ru/C, respectively.
It is well noted that downsizing the metal particles that are supported on the (hydr)oxide supports is conducive to magnifying the interfacial effects[227]. However, the (hydr)oxide supports with relatively larger sizes can exhibit lower conductivity, which is unacceptable for electrocatalysis. To this end, a new strategy was developed by creating oxide islands on metal substrate surfaces to form oxide/metal inverse interfaces, which can effectively amplify the interfacial effect under a high conductivity[228,229]. This strategy has been widely used in the CO oxidation and O2 reduction reactions, such as the FeNi(OH)x/Pt[230], TMO1–x/Pt(111) (TM = Fe, Co, Ni)[231], and atomically dispersed RuOx-on-Pd NS catalysts[232]. For alkaline HOR electrocatalysis, Fu et al. reported the synthesis of IrWOx NBs by modifying Ir-W nanocrystallines with amorphous WOx clusters[233]. The HAADF-STEM images clearly show the interfaces between amorphous WOx clusters and the highly crystalline Ir-W twigs, suggesting a strong interaction at the Ir-W/WOx interface [Figure 10G and H]. According to the DFT calculations, the Ir surface decorated with WOx clusters exhibits stronger adsorption ability for OH species, leading to OH-adsorbed WOx clusters
As reviewed above, the electrocatalytic activities of PGM-based catalysts are closely related to the atomic arrangement, size, dispersion, dimension, component, active layer, and interface. [Table 1] summarizes the specific activities and mass activities of various types of PGM-based electrocatalysts in alkaline HOR electrocatalysis in recent years[25,60,75,109,117,137,153,164,177,178,185,186,209,217,222,224,226,233-258]. Their electrocatalytic performance can be improved by several strategies, including the design of metastable or amorphous nanoscale catalysts to expose abundant active sites, introducing foreign metal atoms to form multicomponent catalysts towards optimizing the surface electronic structures, constructing effective internal and/or external interfaces with strong interfacial interactions, etc.
Comparison of alkaline HOR activities of the latest PGM-based electrocatalysts
| Catalyst | Electrolyte | Catalyst loading (μgPGM cmgeo-2) | j0,s (mA cmPGM-2) | jk,m (A mgPGM-1) @50 mV | Ref. |
| Pt6 NCs/C | 0.1 M KOH | 5 | 1.546 | 3.658 | [137] |
| Mo-Pt/NC | 0.1 M KOH | 10 | 6.37 | 4.549 | [234] |
| Pt1@Co1 CN | 0.1 M KOH | 11.2 | - | 0.68 | [235] |
| Pt-MoC@NC | 0.1 M KOH | 10 | 0.56 | 0.833 | [236] |
| Acid-PtNi/C | 0.1 M KOH | 10 | 1.89 | 0.474 | [75] |
| d-Rh2P NWs | 0.1 M KOH | 10.2 | 7.85 | [209] | |
| RhMo | 0.1 M KOH | 17.6 | - | 6.96 | [164] |
| Rh2Sb NBs/C | 0.1 M KOH | 6.38 | 0.506 | 3.254 | [237] |
| Rh/C-2 | 0.1 M KOH | 3.5 | 0.93 | 0.68 | [238] |
| RhSn/C | 0.1 M KOH | 5.5 | 0.933 | 0.59 | [239] |
| Rh NP/PC | 0.1 M KOH | 54.8 | 0.164 | 0.163 | [240] |
| Rh-Rh2O3-NPs/C | 0.1 M KOH | 28 | 0.425 | 0.119 | [222] |
| Rh2P/C | 0.1 M KOH | 6.4 | 0.65 | 0.52 | [60] |
| P-Rh/C | 0.1 M KOH | 19.23 | 0.494 | 0.292 (@10 mV) | [241] |
| P-Ru/C | 0.1 M KOH | 6.06 | 0.72 | 0.43 | [242] |
| Ru@TiO2 | 0.1 M KOH | 25 | - | 0.275 | [226] |
| IO-Ru-TiO2/C | 0.1 M KOH | 25.48 | 0.11 | 0.907 | [243] |
| Mo-Ru-2/C | 0.1 M KOH | 6 | 0.35 | 1.86 | [244] |
| RuP/NOC | 0.1 M KOH | 18.1 | - | 3.25 | [245] |
| Ru/PEI-XC | 0.1 M KOH | 21.7 | 0.687 | 0.423 | [246] |
| RuNi1 | 0.1 M KOH | 8.8 | - | 2.7 | [185] |
| MoOx-Ru | 0.1 M KOH | 1.0 | 1.66 | 5.72 | [109] |
| Ru7Ni3/C | 0.1 M KOH | 3.9 | - | 9.4 | [247] |
| IrNi@Ir/C | 0.1 M KOH | 10 | 1.22 | 1.12 | [248] |
| IrMo0.59 NPs | 0.1 M KOH | 30 | 1.15 | 2.80 | [178] |
| Ir/Ni–NiO/CNT | 0.1 M KOH | 29.46 | 0.145 | 1.59 | [249] |
| Ir/MoS2 | 0.1 M KOH | 200 μgtotal cmgeo-2 | 1.28 | 0.182 | [250] |
| Ir ONA | 0.1 M KOH | 30 | 0.518 | - | [25] |
| IrWOx/C | 0.1 M KOH | 2.01 | 1.56 | 2.16 | [233] |
| Pd-Pd4S/C | 0.1 M KOH | 8.312 | 0.225 | 37.25 (@10 mV) | [251] |
| 0.38 CeOx-Pd/C | 0.1 M KOH | 13 | 0.118 | 0.515 (@100 mV) | [224] |
| IrNi@PdIr/C | 0.1 M KOH | 19.7 | 0.209 | 0.854 | [252] |
| PdCu/C-500℃ | 0.1 M KOH | 12.5 | 0.833 | 0.522 | [253] |
| Pt2-Rh NSs | 0.1 M KOH | 15.3 | 7.15 | 9.61 | [186] |
| Rh@Pt0.83 NBs | 1 M KOH | 15.3 | 0.592 mA cmPt-2 | 0.214 A mgPt-1 | [217] |
| Ru-Ir(2/3)/C | 0.1 M NaOH | 10 | 0.283 | 0.210 | [177] |
| PtPdCu1/C | 0.1 M KOH | 28.3 | - | 0.6 | [254] |
| PtRu/Mo2C-TaC | 0.1 M KOH | 13 | 0.20 | 0.403 | [255] |
| Ru-increased PtRuX/PC NPs | 0.1 M KOH | 4 | - | 4.4 | [256] |
| a-Pt53Ru47 NWs/C | 0.1 M KOH | 10.2 | 1.90 | 13.7 (@25 mV) | [117] |
| PtRh NAA | 0.1 M KOH | 25 | 1.25 | 0.322 | [257] |
| PtIr DNWs/C | 0.1 M KOH | 10.2 | 0.72 mA | 3.48 | [153] |
| HEA SNWs/C | 0.1 M KOH | 8.8 | - | 6.75 | [258] |
FUEL CELL PERFORMANCES
In most cases, high-performance PGM-based electrocatalysts measured under experimental conditions may not necessarily achieve ideal fuel cell performances due to their different testing conditions. For the rotating disk electrode (RDE) testing system, the electrocatalysts are performed in aqueous electrolytes to achieve a better control of mass and electron transfer, which is conducive to the exploration of structure-activity relationships. However, the aqueous electrolyte is not applicable for AEMFCs that possess solid polymer electrolytes, also known as anion exchange membranes (AEM)[4,259]. In alkaline aqueous electrolytes, the H2 reactants arriving at the catalytic reaction interface are easily affected by the aqueous solution environment, including the electric double layer, water structure, ion effects, etc. This condition is different from the testing system of fuel cells, which generally possess a porous catalytic layer with abundant catalytic interfaces. The porous catalytic layer is instrumental in the direct electrocatalytic reaction between H2 and active interfaces, indicating that the AEMFCs exhibit a faster mass transfer of H2 than the RDE testing system[260]. Meanwhile, the molecular constituents of AEM, such as phenyl and ionic groups, may interact with active species and, thus, influence the performance of AEMFCs. Chung et al. used spectroscopic techniques to detect the co-adsorption behavior of quaternary ammonium cation-hydroxide-water on the catalytic surface of Pt catalysts, where the characteristic peak of tetramethylammonium cation evolved with time, accompanied by the cumulation of the characteristic peaks of hydroxide and water[261]. The cumulative co-adsorption behavior can block the active centers and undoubtedly inhibit the activity of AEMFCs. For the first time, this group further investigated the impact of phenyl group adsorption abilities on the HOR performance of Pt. They found that reducing the adsorption ability of phenyl groups can improve the PPD by three times in AEMFCs[262]. More recently, Maurya et al. investigated the adsorption behaviors of Pt-based and Pd-based catalysts involving Pt/C and Pt-Ru/C, Pd/C, and Pd/C-CeO2, respectively, by combining RDE, DFT calculations, and AEMFC performance tests[263]. They found that the surface adsorption behavior for ionomer components is closely related to the AEMFC performance and stability. The strong hydrogen absorption ability may prevent the co-adsorption of cation-hydroxide-water on Pd-based catalysts, which is conducive to HOR processes in AEMFCs, yet the easy hydrogenation of phenyl groups adsorbed on the surface can conversely reduce the activity. These results indicate that the interaction at the catalyst-electrolyte interface in AEMFC electrodes is another significant factor in affecting the electrocatalytic performance.
Generally, the AEMFCs can achieve a larger limiting current density (> 1 A cm–2), whereas a large product may not be excluded timely if under such a large current density, resulting in a flooding phenomenon that will increase the resistance for mass and electron transfer[264-266]. As a result, water flooding is also a key issue that strictly influences the performance instability of AEMFCs, which makes water management within the cell a prerequisite of high-performance AEMFCs. In the past several years, numerous efforts have been dedicated to optimizing the properties of various components to improve the water management of AEMFCs, including the gas diffusion layers, electrodes, and solid polymer electrolytes[266]. Since the activation operation conducted in the RDE system is different from that in the AEMFC single-cell system, the catalysts with excellent electrocatalytic activity in the former may not exhibit satisfying performance in the latter[267]. Consequently, sufficient research should be conducted on the electrocatalytic performance of HOR in AEMFCs to facilitate the screening of high-performance catalysts for future research. So far, besides the HOR electrocatalysis, the application of PGM-based catalysts in AEMFC single-cell performance has also been provided. For example, Qin et al. prepared core-shell IrNi@PdIr/C anode catalysts for electrocatalyzing AEMFC with a maximum PPD of 311 mW cm-2, much higher than that of Pt/C anodes (233 mW cm-2)[252]. Alesker et al. reported that the PPD of an AEMFC based on a Pd/Ni anode catalyst was much higher than that of the same cell based on a pure Pd catalyst (400 mW cm-2vs. 180 mW cm-2)[268]. The Pd-based anode catalysts with CeO2 modification assembled in AEMFCs were reported to exhibit significantly higher activities and stabilities than the pure Pd/C[223,224,269,270], where the CeO2-modified Pd-based catalysts deliver the peak power densities from 460 to 1,000 mW cm-2. Moreover, Xue et al. reported that the AEMFC with Ru7Ni3/C anodes exhibited an extremely high PPD of 2.03 W cm−2 and excellent durability for over 100 h of operation, superior to the PtRu/C anode catalyst[247]. Table 2 summarizes the AEMFC single-cell performance data from recently reported PGM-based anode catalysts[77,143,164,207,223,224,247,252,255,268,269,271-285]. It could be seen that the PPD of AEMFC has improved from 120 mW cm-2 for 11 nm Ru/C anodes in 2013 to 2150 mW cm-2 for Pt0.25Ru0.75/pN-C anodes in 2023, demonstrating a significant development of PGM-based electrocatalysts in AEMFC applications. It should be noted that the loading amounts of precious metals should be strictly controlled to maintain appropriate cost-effectiveness, though the AEMFC performance is also related to many other conditions.
Comparison of AEMFC performances using recently reported PGM-based electrocatalysts
| Anode | Cathode | T (°C) | Back pressure | PPD (W cm-2) | j@PPD (A cm-2) | Ref. | ||
| Catalyst | Loading (mgPGM cm-2) | Catalyst | Loading (mgPGM cm-2) | |||||
| PtRu/C | 0.4 | Pt/C | 0.4 | 60 | H2-O2 0.1MPa | 1 | 2.2 | [271] |
| Pt/C | 0.6 | 1.5 | ||||||
| Pt-Ru/C | 0.4 | Pt/C | 0.4 | 80 | H2-O2 0.1MPa | 1.85 | 4 | [272] |
| Pt/C | 1.7 | 4 | ||||||
| Pt0.25Ru0.75/N-C | 0.2 | Pt/C | 0.4 | 80 | H2-O2 noBP | 0.831 | 2.1 | [273] |
| Pt0.25Ru0.75/pN-C | 0.18 | Pt/C | 0.55 | 80 | H2-O2 noBP | 2.15 | 5 | [274] |
| H2-air noBP | 1.05 | 2.8 | ||||||
| 0.16 | Fe-N-C | 1.34 | H2-O2 noBP | 1.46 | 3.4 | |||
| H2-air noBP | 0.754 | - | ||||||
| Pt-RuO2/C | 0.025Pt | Pt/C | 0.6 | 80 | H2-O2 285kPa | 0.77 | 1.4 | [275] |
| PtRu/Mo2C-TaC | 0.7 | Pt/C | 0.7 | 70 | H2 30psi, O2 15psi | 1.2 | 2.2 | [255] |
| Pseudomorphic-Pt@IrPd/C | 0.1 | Pt/C | 0.4 | 80 | H2-O2 0.2MPa | 1.27 | 3 | [276] |
| H2-air (CO-free) 0.2MPa | 0.95 | 2.5 | ||||||
| Ir1Ru1 nanowires/C | 0.1 | Pt/C | 0.3 | 60 | H2-O2 0.1MPa | 0.485 | 1 | [277] |
| IrNi@PdIr/C | 0.1 | Pt/C | 0.3 | 60 | H2-O2 noBP | 0.311 | 0.75 | [252] |
| Pd0.33Ir0.67/N-C | 0.2 | Pt/C | 0.3 | 79 | H2-O2 0.1MPa | 0.514 | 1.25 | [77] |
| Pd/Ni | 0.3 | Ag-based | 3 | 73 | H2 0.4Mpa, air (CO-free) 0.2MPa | 0.4 | 1.3 | [268] |
| Pd/C-CeO2 | 0.3 | Ag-based | 3 | 73 | H2 3barg, air (CO<10ppm) 1barg | 0.5 | 1.5 | [223] |
| Pd-CeO2/C | 0.25 | Pt/C | 0.4 | 80 | H2-O2 noBP | 2 | 5 | [278] |
| Pd/C | 0.25 | 1.3 | 3.9 | |||||
| Ag-Co/C | 0.75Ag | 1 | 3.6 | |||||
| Fe/C | 0.03Fe | 0.96 | 2.7 | |||||
| Pd-CeO2/C | 0.42 | PdCu/C | 0.58 | 70 | H2 3bar, O2 2bar | 1 | 2.5 | [269] |
| CeOx-Pd/C | 0.55 | Pt/C | 0.7 | 60 | H2-O2 noBP | 0.643 | 1.4 | [224] |
| Pd-CeO2/OLC | 0.15 | Pt/C | 0.4 | 80 | H2-O2 noBP | 1 | 3 | [279] |
| Pd-CeO2/CB | 0.9 | 2.2 | ||||||
| Pd/OLC | 0.6 | 1.7 | ||||||
| Pd/CB | 0.05 | 0.1 | ||||||
| RhMo nanosheets/C | 0.2 | Pt/C | 0.2 | H2-O2 | 1.52 0.85 | 3.2 | [164] | |
| H2-air | 1.6 | |||||||
| 3 nm Ru/C | 0.5 | Pt/C | 0.5 | 50 | H2-O2 0.1MPa | 0.25 | 0.6 | [280] |
| 11 nm Ru/C | 0.12 | 0.35 | ||||||
| Ru/Meso C | 0.1 | Pt/C | 0.45 | 80 | H2 0.4MPa, O2 0.2MPa | 1.02 | 2.5 | [281] |
| RuPdIr/C | 0.2 | Ag-based | 1 | 80 | H2-O2 0.1MPa | 0.95 | 2.7 | [282] |
| RuCr | 0.4metal | Pt/C | 0.4 | 80 | H2-O2 0.2MPa | 0.77 | 1.51 | [283] |
| RuFe | 0.4metal | Pt/C | 0.4 | 80 | H2-O2 0.2MPa | 1.44 | 4 | [284] |
| Ru7Ni3/C | 0.2 | Pt/C | 0.4 | 95 | H2-O2 250kPag | 2.03 | 5 | [247] |
| H2-air (CO-free) 250kPag | 1.23 | 2.5 | ||||||
| Ru-Ni diatomic sites/NC | 1total | Pt/C | 0.2 | 80 | H2-O2 0.2MPa | 0.54 | 1.7 | [143] |
| B-Ru/C | 0.42 | Pt/C | 0.48 | 95.5 | H2-O2 0.25MPa | 1.37 | 3 | [285] |
| H2-air (CO-free) 0.25MPa | 0.83 | 1.8 | ||||||
| Ru2P/C | 0.4 | Pt/C | 0.4 | 80 | H2-O2 0.1MPa | 1.3 | 3 | [207] |
CONCLUSION AND OUTLOOK
Developing high-performance PGM-based electrocatalysts for alkaline HOR is conducive to the large-scale commercialization of AEMFCs. However, the HOR performance is always hindered by the sluggish reaction kinetics in alkaline media, making the electrocatalytic reaction mechanism a prerequisite for catalyst design. Thermodynamically, HBE is a pivotal descriptor of the HOR activity, where the strength of metal–H bonds (ΔGH) is related to its inherent properties. According to the elementary steps and experimental conditions in alkaline HOR electrocatalysis, the reaction rate should also be related to the OHBE and subject to the electric double layer, water structure, ion effects, etc. Although still controversial, these activity descriptors have been widely validated after decades of extensive efforts, providing significant assistance in guiding the design of high-performance catalysts. Given the low reserves and high prices of PGMs, novel PGM-based catalysts with high utilization of precious metal atoms have been developed with significantly improved activity via various feasible strategies in the last decade. The electrocatalytic activities are closely related to the atomic arrangement, size/morphology, ligand/strain/ensemble effect, and oxophilicity, all of which have different influences on the surface electronic structures, type, and number of active sites. For PGM-based catalysts, the alkaline HOR electrocatalytic performance can be improved by the following several aspects: (1) designing low-dimensional catalysts with a metastable or amorphous phase that exposes abundantly accessible polytype sites on the surface; (2) optimizing the surface electronic structures or d-band centers of active sites by regulating the type and composition of foreign metal atoms; (3) introducing abundant dual-active sites or atomically distributed diatomic sites for adsorbing Had and OHad intermediates separately and precisely regulating their microstructures and compositions to optimize the electronic and oxophilic effect; (4) designing well-defined intermetallics to precisely regulate the electron interaction and stability; (5) constructing effective internal and/or external interfaces with strong interfacial interactions in heterostructures to optimize the electron structures. These useful and attractive strategies are very helpful in improving the performance of PGM-based anode catalysts in AEMFC. Despite recent breakthroughs in the preparation of alkaline HOR electrocatalysts based on PGM materials, some challenges and the potential outlook in the insight of catalytic mechanisms and practical applications of catalytic materials have been proposed.
It is still difficult to conclude a universal perception of the alkaline HOR catalytic mechanism due to the complicated catalytic environment in alkaline electrolytes. Thermodynamically, we can explain the catalytic reaction process based on the ΔGH and ΔGOH. However, the adsorption/desorption behavior of Had and OHad intermediates is a dynamic recycling process that involves the impact of increased coverage of Had and OHad (θH > 0; θOH > 0) on the electrocatalytic surface. Shinagawa et al. have concluded that the various θH could lead to different Tafel slopes, suggesting a constantly changing catalytic reaction rate on the electrode surface[37]. Thus, when using theoretical methods to calculate ΔGH and ΔGOH, the catalyst model cannot be simply placed in a vacuum environment for calculation but rather needs to consider the θH and θOH and more practical testing conditions. The theoretical exploration cannot fully explain experimental results and more comprehensive and direct experimental evidence needs to be combined to obtain widespread recognition. Owing to the difficulty in real-time monitoring of the microenvironment in alkaline electrolytes, the real active centers and the adsorption/desorption behavior of reaction intermediates may not be accurately judged. Even worse, if a catalyst is not stable enough, the surface oxidation or reduction that occurs during the electrocatalytic process may alter the type of active sites. Due to the stable surface structure of PGMs (especially Pt) during the electrocatalytic process, most of these mechanism conclusions are obtained by using them as model catalysts. However, for multi-component catalysts, especially nanomaterials containing non-precious metals, their much stronger oxygen oxophilicity may lead to irreversible oxidation during the HOR process, such as Ni, Mo, W, etc. Therefore, this type of catalyst may exhibit different reaction pathways. To get insight into the real active centers and reaction intermediates, advanced in situ operando characterization techniques, including in situ Raman spectroscopy, infrared spectroscopy, ambient pressure XPS, and XAS, and the isotope experiments[286,287] are conducted to monitor and trace their dynamic behaviors in recent years of research, providing experimental and theoretical basis for the further exploration of mechanisms.
In terms of the design of PGM-based catalysts, although researchers have explored many attractive methods to reduce the loading of PGMs while improving their overall catalytic performance, their high prices still limit the large-scale application of AEMFCs. Moreover, for the applications of AEMFCs in watts to thousands of watts, the catalytic activity of alkaline anode catalysts at high current densities seems no longer to be a critical factor because the performance of AEMFCs is also closely related to many other factors, such as the constituent of AEM polymeric electrolytes, co-adsorption of cation-hydroxide-water, and water management. In recent years, PGM-free catalysts, including Ni/Co-based alloys/nitrides/phosphides and Mo/W-based carbides, have been investigated to show excellent performance in electrocatalyzing alkaline HOR, implying a potential application in the anode catalysts of AEMFCs[288-290]. Especially, the single-cell performance can achieve peak power densities from ~120 to ~628 mW cm−2 when assembled with Ni-based anode catalysts, such as NiMo alloys, Ni@CNx, and Ni-H2-NH3 anodes[291-293]. Although the performance of AEMFC with Ni-based anode catalysts still lags far behind that of the same cell based on PGM-based anode catalysts, the development of PGM-free AEMFCs continues to hold great potential.
DECLARATIONS
Authors’ contributions
Prepared and revised the manuscript: Fu L
Revised the manuscript: Wang S, Cai J, Huang H
Methodology and supervision: Yang F
Supervision, funding acquisition, and writing-review & editing: Xie S
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant Nos. 22171093, 22201085, and 22202172); the Natural Science Foundation of Fujian Province (Grant Nos. 2022J05058 and 2022J02008); the Fundamental Research Funds for the Central Universities (Grant No. ZQN-1106); and the Fujian Provincial Chemistry Discipline Alliance Foundation.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Staffell I, Scamman D, Velazquez Abad A, et al. The role of hydrogen and fuel cells in the global energy system. Energy Environ Sci 2019;12:463-91.
2. Wang Y, Chen KS, Mishler J, Cho SC, Adroher XC. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Applied Energy 2011;88:981-1007.
3. Meyer Q, Zeng Y, Zhao C. In situ and operando characterization of proton exchange membrane fuel cells. Adv Mater 2019;31:e1901900.
4. Ramaswamy N, Mukerjee S. Alkaline anion-exchange membrane fuel cells: challenges in electrocatalysis and interfacial charge transfer. Chem Rev 2019;119:11945-79.
5. Yang Y, Peltier CR, Zeng R, et al. Electrocatalysis in alkaline media and alkaline membrane-based energy technologies. Chem Rev 2022;122:6117-321.
6. Wang XX, Swihart MT, Wu G. Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation. Nat Catal 2019;2:578-89.
7. Li Z, Wang Y, Mu Y, et al. Recent advances in the anode catalyst layer for proton exchange membrane fuel cells. Renew Sustain Energy Rev 2023;176:113182.
8. Wang Z, Sankarasubramanian S, Ramani V. Advances in anion exchange membranes for electrochemical energy conversion. Curr Opin Electrochem 2018;12:240-5.
9. Noh S, Jeon JY, Adhikari S, Kim YS, Bae C. Molecular engineering of hydroxide conducting polymers for anion exchange membranes in electrochemical energy conversion technology. Acc Chem Res 2019;52:2745-55.
10. Gao FY, Gao MR. Nickel-based anode catalysts for efficient and affordable anion-exchange membrane fuel cells. Acc Chem Res 2023;56:1445-57.
11. Yang F, Tian X, Luo W, Feng L. Alkaline hydrogen oxidation reaction on Ni-based electrocatalysts: From mechanistic study to material development. Coordination Chemistry Reviews 2023;478:214980.
12. Luo F, Wagner S, Onishi I, et al. Surface site density and utilization of platinum group metal (PGM)-free Fe-NC and FeNi-NC electrocatalysts for the oxygen reduction reaction. Chem Sci 2020;12:384-96.
13. Dai L, Xue Y, Qu L, Choi HJ, Baek JB. Metal-free catalysts for oxygen reduction reaction. Chem Rev 2015;115:4823-92.
14. Gong K, Du F, Xia Z, Durstock M, Dai L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 2009;323:760-4.
15. Mu X, Liu S, Chen L, Mu S. Alkaline hydrogen oxidation reaction catalysts: insight into catalytic mechanisms, classification, activity regulation and challenges. Small Structures 2023;4:2200281.
16. Davydova ES, Mukerjee S, Jaouen F, Dekel DR. Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal 2018;8:6665-90.
17. Durst J, Siebel A, Simon C, Hasché F, Herranz J, Gasteiger HA. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ Sci 2014;7:2255-60.
18. Cong Y, Yi B, Song Y. Hydrogen oxidation reaction in alkaline media: From mechanism to recent electrocatalysts. Nano Energy 2018;44:288-303.
19. Yao ZC, Tang T, Jiang Z, Wang L, Hu JS, Wan LJ. Electrocatalytic hydrogen oxidation in alkaline media: from mechanistic insights to catalyst design. ACS Nano 2022;16:5153-83.
20. Parsons R. The rate of electrolytic hydrogen evolution and the heat of adsorption of hydrogen. Trans Faraday Soc 1958;54:1053-63.
21. Nørskov JK, Bligaard T, Logadottir A, et al. Trends in the exchange current for hydrogen evolution. J Electrochem Soc 2005;152:J23.
22. Seh ZW, Kibsgaard J, Dickens CF, Chorkendorff I, Nørskov JK, Jaramillo TF. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017;355:eaad4998.
23. Ohyama J, Sato T, Yamamoto Y, Arai S, Satsuma A. Size specifically high activity of Ru nanoparticles for hydrogen oxidation reaction in alkaline electrolyte. J Am Chem Soc 2013;135:8016-21.
24. Lee W, Bera S, Woo H, et al. Controllable size and crystallinity of Ru nanoparticles on a carbon support synthesized by fluidized bed reactor-atomic layer deposition for enhanced hydrogen oxidation activity. J Mater Chem A 2021;9:17223-30.
25. Yang F, Fu L, Cheng G, Chen S, Luo W. Ir-oriented nanocrystalline assemblies with high activity for hydrogen oxidation/evolution reactions in an alkaline electrolyte. J Mater Chem A 2017;5:22959-63.
26. Zhu J, Chen Z, Xie M, et al. Iridium-based cubic nanocages with 1.1-nm-thick walls: a highly efficient and durable electrocatalyst for water oxidation in an acidic medium. Angew Chem Int Ed Engl 2019;58:7244-8.
27. Bu L, Ding J, Guo S, et al. A general method for multimetallic platinum alloy nanowires as highly active and stable oxygen reduction catalysts. Adv Mater 2015;27:7204-12.
28. Wang P, Jiang K, Wang G, Yao J, Huang X. Phase and interface engineering of platinum-nickel nanowires for efficient electrochemical hydrogen evolution. Angew Chem Int Ed Engl 2016;55:12859-63.
29. Scofield ME, Zhou Y, Yue S, et al. Role of chemical composition in the enhanced catalytic activity of pt-based alloyed ultrathin nanowires for the hydrogen oxidation reaction under alkaline conditions. ACS Catal 2016;6:3895-908.
30. An L, Zhao X, Zhao T, Wang D. Atomic-level insight into reasonable design of metal-based catalysts for hydrogen oxidation in alkaline electrolytes. Energy Environ Sci 2021;14:2620-38.
31. Liang J, Ma F, Hwang S, et al. Atomic arrangement engineering of metallic nanocrystals for energy-conversion electrocatalysis. Joule 2019;3:956-91.
32. Zhao R, Yue X, Li Q, Fu G, Lee JM, Huang S. Recent advances in electrocatalysts for alkaline hydrogen oxidation reaction. Small 2021;17:e2100391.
33. Gottesfeld S, Dekel DR, Page M, et al. Anion exchange membrane fuel cells: Current status and remaining challenges. J Power Sources 2018;375:170-84.
34. Mustain WE. Understanding how high-performance anion exchange membrane fuel cells were achieved: Component, interfacial, and cell-level factors. Curr Opin Electrochem 2018;12:233-9.
35. Li D, Chung HT, Maurya S, Matanovic I, Kim YS. Impact of ionomer adsorption on alkaline hydrogen oxidation activity and fuel cell performance. Curr Opin Electrochem 2018;12:189-95.
36. Zeradjanin AR, Vimalanandan A, Polymeros G, Topalov AA, Mayrhofer KJJ, Rohwerder M. Balanced work function as a driver for facile hydrogen evolution reaction - comprehension and experimental assessment of interfacial catalytic descriptor. Phys Chem Chem Phys 2017;19:17019-27.
37. Shinagawa T, Garcia-Esparza AT, Takanabe K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci Rep 2015;5:13801.
38. Shi Y, Zhang B. Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction. Chem Soc Rev 2016;45:1529-41.
39. Zheng J, Sheng W, Zhuang Z, Xu B, Yan Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci Adv 2016;2:e1501602.
40. Sheng W, Gasteiger HA, Shao-horn Y. Hydrogen oxidation and evolution reaction kinetics on platinum: acid vs alkaline electrolytes. J Electrochem Soc 2010;157:B1529.
41. Tian X, Zhao P, Sheng W. Hydrogen evolution and oxidation: mechanistic studies and material advances. Adv Mater 2019;31:e1808066.
42. Elbert K, Hu J, Ma Z, et al. Elucidating hydrogen oxidation/evolution kinetics in base and acid by enhanced activities at the optimized pt shell thickness on the Ru core. ACS Catal 2015;5:6764-72.
43. Montero MA, de Chialvo MRG, Chialvo AC. Evaluation of the kinetic parameters of the hydrogen oxidation reaction on nanostructured iridium electrodes in alkaline solution. J Electroanal Chem 2016;767:153-9.
44. Montero MA, Gennero de Chialvo MR, Chialvo AC. Kinetics of the hydrogen oxidation reaction on nanostructured rhodium electrodes in alkaline solution. J Power Sources 2015;283:181-6.
45. Rheinländer PJ, Herranz J, Durst J, Gasteiger HA. Kinetics of the hydrogen oxidation/evolution reaction on polycrystalline platinum in alkaline electrolyte reaction order with respect to hydrogen pressure. J Electrochem Soc 2014;161:F1448-57.
46. Campos-roldán CA, Alonso-vante N. The hydrogen oxidation reaction in alkaline medium: an overview. Electrochem Energ Rev 2019;2:312-31.
47. Trasatti S. Work function, electronegativity, and electrochemical behaviour of metals: III. electrolytic hydrogen evolution in acid solutions. J Electroanal Chem Interfacial Electrochem 1972;39:163-84.
48. Bligaard T, Nørskov J, Dahl S, Matthiesen J, Christensen C, Sehested J. The brønsted–evans–polanyi relation and the volcano curve in heterogeneous catalysis. Journal of Catalysis 2004;224:206-17.
49. Sheng W, Myint M, Chen JG, Yan Y. Correlating the hydrogen evolution reaction activity in alkaline electrolytes with the hydrogen binding energy on monometallic surfaces. Energy Environ Sci 2013;6:1509.
50. Sheng W, Zhuang Z, Gao M, Zheng J, Chen JG, Yan Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat Commun 2015;6:5848.
51. Zhuang L, Jin J, Abruña HD. Direct observation of electrocatalytic synergy. J Am Chem Soc 2007;129:11033-5.
52. van der Niet MJ, Garcia-araez N, Hernández J, Feliu JM, Koper MT. Water dissociation on well-defined platinum surfaces: the electrochemical perspective. Catalysis Today 2013;202:105-13.
53. Zheng J, Nash J, Xu B, Yan Y. Perspective-towards establishing apparent hydrogen binding energy as the descriptor for hydrogen oxidation/evolution reactions. J Electrochem Soc 2018;165:H27-9.
54. Giles SA, Wilson JC, Nash J, Xu B, Vlachos DG, Yan Y. Recent advances in understanding the pH dependence of the hydrogen oxidation and evolution reactions. Journal of Catalysis 2018;367:328-31.
55. Strmcnik D, Uchimura M, Wang C, et al. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat Chem 2013;5:300-6.
56. Alia SM, Pivovar BS, Yan Y. Platinum-coated copper nanowires with high activity for hydrogen oxidation reaction in base. J Am Chem Soc 2013;135:13473-8.
57. Ramaswamy N, Ghoshal S, Bates MK, Jia Q, Li J, Mukerjee S. Hydrogen oxidation reaction in alkaline media: Relationship between electrocatalysis and electrochemical double-layer structure. Nano Energy 2017;41:765-71.
58. Wang YH, Wang XT, Ze H, et al. Spectroscopic verification of adsorbed hydroxy intermediates in the bifunctional mechanism of the hydrogen oxidation reaction. Angew Chem Int Ed Engl 2021;60:5708-11.
59. Liao J, Ding W, Tao S, et al. Carbon supported IrM (M = Fe, Ni, Co) alloy nanoparticles for the catalysis of hydrogen oxidation in acidic and alkaline medium. Chin J Catal 2016;37:1142-8.
60. Yang F, Bao X, Gong D, et al. Rhodium phosphide: a new type of hydrogen oxidation reaction catalyst with non-linear correlated catalytic response to pH. ChemElectroChem 2019;6:1990-5.
61. Cong Y, Meng F, Wang X, et al. Uniform PtRu0.6 nanoparticles supported on nitrogen-doped carbon obtained from ZIF-8/GO hybrid with remarkable alkaline hydrogen oxidation activity. J Electron Mater 2023;52:2388-95.
62. Subbaraman R, Tripkovic D, Strmcnik D, et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011;334:1256-60.
63. Danilovic N, Subbaraman R, Strmcnik D, et al. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew Chem Int Ed Engl 2012;51:12495-8.
64. Subbaraman R, Tripkovic D, Chang KC, et al. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat Mater 2012;11:550-7.
65. Mccrum IT, Koper MTM. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat Energy 2020;5:891-9.
66. Ledezma-yanez I, Wallace WDZ, Sebastián-pascual P, Climent V, Feliu JM, Koper MTM. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat Energy 2017;2:17031.
67. Rebollar L, Intikhab S, Oliveira NJ, et al. “Beyond adsorption” descriptors in hydrogen electrocatalysis. ACS Catal 2020;10:14747-62.
68. Sarabia FJ, Sebastián-Pascual P, Koper MTM, Climent V, Feliu JM. Effect of the interfacial water structure on the hydrogen evolution reaction on Pt(111) modified with different nickel hydroxide coverages in alkaline media. ACS Appl Mater Interfaces 2019;11:613-23.
69. Chen X, McCrum IT, Schwarz KA, Janik MJ, Koper MTM. Co-adsorption of cations as the cause of the apparent pH dependence of hydrogen adsorption on a stepped platinum single-crystal electrode. Angew Chem Int Ed Engl 2017;56:15025-9.
70. Liu E, Li J, Jiao L, et al. Unifying the hydrogen evolution and oxidation reactions kinetics in base by identifying the catalytic roles of hydroxyl-water-cation adducts. J Am Chem Soc 2019;141:3232-9.
71. Zhang M, de Respinis M, Frei H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat Chem 2014;6:362-7.
72. Kornienko N, Resasco J, Becknell N, et al. Operando spectroscopic analysis of an amorphous cobalt sulfide hydrogen evolution electrocatalyst. J Am Chem Soc 2015;137:7448-55.
73. Feng Z, Li L, Zheng X, et al. Role of hydroxyl species in hydrogen oxidation reaction: a DFT study. J Phys Chem C 2019;123:23931-9.
74. Li P, Jiang Y, Hu Y, et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat Catal 2022;5:900-11.
75. Lu S, Zhuang Z. Investigating the influences of the adsorbed species on catalytic activity for hydrogen oxidation reaction in alkaline electrolyte. J Am Chem Soc 2017;139:5156-63.
76. Shen L, Lu B, Qu X, et al. Does the oxophilic effect serve the same role for hydrogen evolution/oxidation reaction in alkaline media? Nano Energy 2019;62:601-9.
77. Cong Y, Mccrum IT, Gao X, et al. Uniform Pd 0.33 Ir 0.67 nanoparticles supported on nitrogen-doped carbon with remarkable activity toward the alkaline hydrogen oxidation reaction. J Mater Chem A 2019;7:3161-9.
78. Jang SW, Dutta S, Kumar A, et al. Holey Pt nanosheets on NiFe-hydroxide laminates: synergistically enhanced electrocatalytic 2D interface toward hydrogen evolution reaction. ACS Nano 2020;14:10578-88.
79. Wang Y, Chen L, Yu X, Wang Y, Zheng G. Superb alkaline hydrogen evolution and simultaneous electricity generation by pt-decorated Ni 3 N nanosheets. Adv Energy Mater 2017;7:1601390.
80. Xia Y, Xiong Y, Lim B, Skrabalak SE. Shape-controlled synthesis of metal nanocrystals: simple chemistry meets complex physics? Angew Chem Int Ed Engl 2009;48:60-103.
81. Markovic N, Ross Jr. P. Surface science studies of model fuel cell electrocatalysts. Surface Science Reports 2002;45:117-229.
82. Ma Z, Cano ZP, Yu A, et al. Enhancing oxygen reduction activity of pt-based electrocatalysts: from theoretical mechanisms to practical methods. Angew Chem Int Ed Engl 2020;59:18334-48.
83. Stamenkovic V, Mun BS, Mayrhofer KJ, et al. Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure. Angew Chem Int Ed Engl 2006;45:2897-901.
84. Hammer B, Nørskov J. Electronic factors determining the reactivity of metal surfaces. Surface Science 1995;343:211-20.
85. Marković NM, Grgur BN, Ross PN. Temperature-dependent hydrogen electrochemistry on platinum low-index single-crystal surfaces in acid solutions. J Phys Chem B 1997;101:5405-13.
86. Schmidt T, Ross P, Markovic N. Temperature dependent surface electrochemistry on Pt single crystals in alkaline electrolytes Part 2. The hydrogen evolution/oxidation reaction. J Electroanal Chem 2002;524-5:252-60.
87. Barber J, Conway B. Structural specificity of the kinetics of the hydrogen evolution reaction on the low-index surfaces of Pt single-crystal electrodes in 0.5 M dm−3 NaOH. J Electroanal Chem 1999;461:80-9.
88. Skúlason E, Tripkovic V, Björketun ME, et al. Modeling the electrochemical hydrogen oxidation and evolution reactions on the basis of density functional theory calculations. J Phys Chem C 2010;114:18182-97.
89. Markovića NM, Sarraf ST, Gasteiger HA, Ross PN. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J Chem Soc , Faraday Trans 1996;92:3719-25.
90. Yao Y, He DS, Lin Y, et al. Modulating fcc and hcp ruthenium on the surface of palladium-copper alloy through tunable lattice mismatch. Angew Chem Int Ed Engl 2016;55:5501-5.
91. Cheng H, Yang N, Lu Q, Zhang Z, Zhang H. Syntheses and properties of metal nanomaterials with novel crystal phases. Adv Mater 2018;30:e1707189.
92. Wang X, Figueroa-Cosme L, Yang X, et al. Pt-based icosahedral nanocages: using a combination of {111} facets, twin defects, and ultrathin walls to greatly enhance their activity toward oxygen reduction. Nano Lett 2016;16:1467-71.
93. Chen Y, Fan Z, Luo Z, et al. High-yield synthesis of crystal-phase-heterostructured 4H/fcc Au@Pd core-shell nanorods for electrocatalytic ethanol oxidation. Adv Mater 2017;29:1701331.
94. Li WZ, Liu JX, Gu J, et al. Chemical insights into the design and development of face-centered cubic ruthenium catalysts for fischer-tropsch synthesis. J Am Chem Soc 2017;139:2267-76.
95. Zheng Y, Jiao Y, Zhu Y, et al. High electrocatalytic hydrogen evolution activity of an anomalous ruthenium catalyst. J Am Chem Soc 2016;138:16174-81.
96. Huang JL, Li Z, Duan HH, et al. Formation of hexagonal-close packed (HCP) rhodium as a size effect. J Am Chem Soc 2017;139:575-8.
98. Fan Z, Bosman M, Huang X, et al. Stabilization of 4H hexagonal phase in gold nanoribbons. Nat Commun 2015;6:7684.
99. Zheng H, Cao A, Weinberger CR, et al. Discrete plasticity in sub-10-nm-sized gold crystals. Nat Commun 2010;1:144.
100. Guo Q, Zhao Y, Mao WL, Wang Z, Xiong Y, Xia Y. Cubic to tetragonal phase transformation in cold-compressed Pd nanocubes. Nano Lett 2008;8:972-5.
101. Sun Y, Yang W, Ren Y, Wang L, Lei C. Multiple-step phase transformation in silver nanoplates under high pressure. Small 2011;7:606-11.
102. Wang H, Zhou S, Gilroy KD, Cai Z, Xia Y. Icosahedral nanocrystals of noble metals: Synthesis and applications. Nano Today 2017;15:121-44.
103. Patala S, Marks LD, Olvera de la Cruz M. Thermodynamic analysis of multiply twinned particles: surface stress effects. J Phys Chem Lett 2013;4:3089-94.
104. Vasquez Y, Luo Z, Schaak RE. Low-temperature solution synthesis of the non-equilibrium ordered intermetallic compounds Au3Fe, Au3Co, and Au3Ni as nanocrystals. J Am Chem Soc 2008;130:11866-7.
105. Bondi JF, Misra R, Ke X, Sines IT, Schiffer P, Schaak RE. Optimized synthesis and magnetic properties of intermetallic Au3Fe1-x, Au3Co1-x, and Au3Ni1- x nanoparticles. Chem Mater 2010;22:3988-94.
106. Wang Y, Peng HC, Liu J, Huang CZ, Xia Y. Use of reduction rate as a quantitative knob for controlling the twin structure and shape of palladium nanocrystals. Nano Lett 2015;15:1445-50.
107. Kusada K, Kobayashi H, Yamamoto T, et al. Discovery of face-centered-cubic ruthenium nanoparticles: facile size-controlled synthesis using the chemical reduction method. J Am Chem Soc 2013;135:5493-6.
108. Zhao T, Xiao D, Chen Y, et al. Boosting alkaline hydrogen electrooxidation on an unconventional fcc-Ru polycrystal. J Energy Chem 2021;61:15-22.
109. Li L, Liu C, Liu S, et al. Phase engineering of a ruthenium nanostructure toward high-performance bifunctional hydrogen catalysis. ACS Nano 2022;16:14885-94.
110. Anantharaj S, Noda S. Amorphous catalysts and electrochemical water splitting: an untold story of harmony. Small 2020;16:e1905779.
111. Zhai W, Sakthivel T, Chen F, Du C, Yu H, Dai Z. Amorphous materials for elementary-gas-involved electrocatalysis: an overview. Nanoscale 2021;13:19783-811.
112. Li MX, Sun YT, Wang C, et al. Data-driven discovery of a universal indicator for metallic glass forming ability. Nat Mater 2022;21:165-72.
113. Yao Y, Huang Z, Xie P, et al. Carbothermal shock synthesis of high-entropy-alloy nanoparticles. Science 2018;359:1489-94.
114. He Y, Liu L, Zhu C, et al. Amorphizing noble metal chalcogenide catalysts at the single-layer limit towards hydrogen production. Nat Catal 2022;5:212-21.
115. Phan QT, Poon KC, Sato H. A review on amorphous noble-metal-based electrocatalysts for fuel cells: Synthesis, characterization, performance, and future perspective. Int J Hydrogen Energy 2021;46:14190-211.
116. Wu G, Zheng X, Cui P, et al. A general synthesis approach for amorphous noble metal nanosheets. Nat Commun 2019;10:4855.
117. Wang S, Fu L, Huang H, et al. Local oxidation induced amorphization of 1.5-nm-thick Pt–Ru nanowires enables superactive and CO-tolerant hydrogen oxidation in alkaline media. Adv Funct Materials 2023;33:2304125.
118. Mayrhofer KJ, Blizanac BB, Arenz M, Stamenkovic VR, Ross PN, Markovic NM. The impact of geometric and surface electronic properties of pt-catalysts on the particle size effect in electrocatalysis. J Phys Chem B 2005;109:14433-40.
119. Gasteiger HA, Kocha SS, Sompalli B, Wagner FT. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B Enviro 2005;56:9-35.
120. Zalitis C, Kucernak AR, Sharman J, Wright E. Design principles for platinum nanoparticles catalysing electrochemical hydrogen evolution and oxidation reactions: edges are much more active than facets. J Mater Chem A 2017;5:23328-38.
121. Shao M, Peles A, Shoemaker K. Electrocatalysis on platinum nanoparticles: particle size effect on oxygen reduction reaction activity. Nano Lett 2011;11:3714-9.
122. Shao M, Peles A, Shoemaker K, et al. Enhanced oxygen reduction activity of platinum monolayer on gold nanoparticles. J Phys Chem Lett 2011;2:67-72.
123. Sun Y, Dai Y, Liu Y, Chen S. A rotating disk electrode study of the particle size effects of Pt for the hydrogen oxidation reaction. Phys Chem Chem Phys 2012;14:2278-85.
124. Jaramillo TF, Jørgensen KP, Bonde J, Nielsen JH, Horch S, Chorkendorff I. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2007;317:100-2.
125. Nesselberger M, Ashton S, Meier JC, Katsounaros I, Mayrhofer KJ, Arenz M. The particle size effect on the oxygen reduction reaction activity of Pt catalysts: influence of electrolyte and relation to single crystal models. J Am Chem Soc 2011;133:17428-33.
126. Zheng J, Zhuang Z, Xu B, Yan Y. Correlating hydrogen oxidation/evolution reaction activity with the minority weak hydrogen-binding sites on Ir/C catalysts. ACS Catal 2015;5:4449-55.
127. Liu L, Corma A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem Rev 2018;118:4981-5079.
128. Yang JC, Small MW, Grieshaber RV, Nuzzo RG. Recent developments and applications of electron microscopy to heterogeneous catalysis. Chem Soc Rev 2012;41:8179-94.
129. Cuenya B, Behafarid F. Nanocatalysis: size- and shape-dependent chemisorption and catalytic reactivity. Surf Sci Rep 2015;70:135-87.
130. Choi CH, Kim M, Kwon HC, et al. Tuning selectivity of electrochemical reactions by atomically dispersed platinum catalyst. Nat Commun 2016;7:10922.
131. Yang S, Kim J, Tak YJ, Soon A, Lee H. Single-atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions. Angew Chem Int Ed Engl 2016;55:2058-62.
132. Kawawaki T, Kataoka Y, Hirata M, et al. Creation of high-performance heterogeneous photocatalysts by controlling ligand desorption and particle size of gold nanocluster. Angew Chem Int Ed Engl 2021;60:21340-50.
133. Chakraborty I, Pradeep T. Atomically precise clusters of noble metals: emerging link between atoms and nanoparticles. Chem Rev 2017;117:8208-71.
134. Duchesne PN, Li ZY, Deming CP, et al. Golden single-atomic-site platinum electrocatalysts. Nat Mater 2018;17:1033-9.
135. Yuan SF, Xu CQ, Liu WD, Zhang JX, Li J, Wang QM. Rod-shaped silver supercluster unveiling strong electron coupling between substituent icosahedral units. J Am Chem Soc 2021;143:12261-7.
136. Yuan P, Zhang R, Selenius E, et al. Solvent-mediated assembly of atom-precise gold-silver nanoclusters to semiconducting one-dimensional materials. Nat Commun 2020;11:2229.
137. Wang X, Zhao L, Li X, et al. Atomic-precision Pt6 nanoclusters for enhanced hydrogen electro-oxidation. Nat Commun 2022;13:1596.
138. Yan H, Lin Y, Wu H, et al. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat Commun 2017;8:1070.
139. Yang J, Li WH, Tan S, et al. The electronic metal-support interaction directing the design of single atomic site catalysts: achieving high efficiency towards hydrogen evolution. Angew Chem Int Ed Engl 2021;60:19085-91.
140. Tang T, Ding L, Jiang Z, Hu J, Wan L. Advanced transition metal/nitrogen/carbon-based electrocatalysts for fuel cell applications. Sci China Chem 2020;63:1517-42.
141. Meng G, Cao H, Wei T, et al. Highly dispersed Ru clusters toward an efficient and durable hydrogen oxidation reaction. Chem Commun 2022;58:11839-42.
142. Zhang Z, Ni L, Liu H, Zhao Z, Yuan X, Li H. Accelerated kinetics of alkaline hydrogen evolution/oxidation reactions on dispersed ruthenium sites through N and S dual coordination. Sci China Chem 2022;65:611-8.
143. Han L, Ou P, Liu W, et al. Design of Ru-Ni diatomic sites for efficient alkaline hydrogen oxidation. Sci Adv 2022;8:eabm3779.
144. Zhang Y, Gao F, You H, Li Z, Zou B, Du Y. Recent advances in one-dimensional noble-metal-based catalysts with multiple structures for efficient fuel-cell electrocatalysis. Coordination Chemistry Reviews 2022;450:214244.
145. Shao Q, Lu K, Huang X. Platinum group nanowires for efficient electrocatalysis. Small Methods 2019;3:1800545.
146. Wang W, Lv F, Lei B, Wan S, Luo M, Guo S. Tuning nanowires and nanotubes for efficient fuel-cell electrocatalysis. Adv Mater 2016;28:10117-41.
147. Zhang G, Jin L, Zhang R, Bai Y, Zhu R, Pang H. Recent advances in the development of electronically and ionically conductive metal-organic frameworks. Coord Chem Rev 2021;439:213915.
148. Zhang M, Xu Y, Wang S, et al. Polyethylenimine-modified bimetallic Au@Rh core–shell mesoporous nanospheres surpass Pt for pH-universal hydrogen evolution electrocatalysis. J Mater Chem A 2021;9:13080-6.
149. Liu K, Wang W, Guo P, et al. Replicating the defect structures on ultrathin Rh nanowires with pt to achieve superior electrocatalytic activity toward ethanol oxidation. Adv Funct Materials 2019;29:1806300.
150. Liu R, Zhao H, Zhao X, et al. Defect sites in ultrathin Pd Nanowires facilitate the highly efficient electrochemical hydrodechlorination of pollutants by H*ads. Environ Sci Technol 2018;52:9992-10002.
151. Huang X, Zhao Z, Chen Y, et al. High density catalytic hot spots in ultrafine wavy nanowires. Nano Lett 2014;14:3887-94.
152. Shi Y, Lyu Z, Zhao M, Chen R, Nguyen QN, Xia Y. Noble-metal nanocrystals with controlled shapes for catalytic and electrocatalytic applications. Chem Rev 2021;121:649-735.
153. Wang M, Wang M, Zhan C, et al. Ultrafine platinum-iridium distorted nanowires as robust catalysts toward bifunctional hydrogen catalysis. J Mater Chem A 2022;10:18972-7.
154. Zhang J, Ye J, Fan Q, et al. Cyclic penta-twinned rhodium nanobranches as superior catalysts for ethanol electro-oxidation. J Am Chem Soc 2018;140:11232-40.
155. Chen Y, Fan Z, Zhang Z, et al. Two-dimensional metal nanomaterials: synthesis, properties, and applications. Chem Rev 2018;118:6409-55.
156. Lyu Z, Zhang X, Liao X, et al. Two-dimensionally assembled Pd–Pt–Ir supernanosheets with subnanometer interlayer spacings toward high-efficiency and durable water splitting. ACS Catal 2022;12:5305-15.
157. Huang X, Tang S, Mu X, et al. Freestanding palladium nanosheets with plasmonic and catalytic properties. Nat Nanotechnol 2011;6:28-32.
158. Kong X, Xu K, Zhang C, et al. Free-standing two-dimensional ru nanosheets with high activity toward water splitting. ACS Catal 2016;6:1487-92.
159. Li Y, Cheng C, Han S, et al. Electrocatalytic reduction of low-concentration nitric oxide into ammonia over Ru nanosheets. ACS Energy Lett 2022;7:1187-94.
160. Ling T, Wang JJ, Zhang H, et al. Freestanding ultrathin metallic nanosheets: materials, synthesis, and applications. Adv Mater 2015;27:5396-402.
161. Jiang B, Guo Y, Kim J, et al. Mesoporous metallic iridium nanosheets. J Am Chem Soc 2018;140:12434-41.
162. Xing Y, Yang Y, Li D, et al. Crumpled Ir nanosheets fully covered on porous carbon nanofibers for long-life rechargeable lithium-CO2 batteries. Adv Mater 2018;30:e1803124.
163. Zhang J, Fan X, Wang S, et al. Surface engineered Ru2Ni multilayer nanosheets for hydrogen oxidation catalysis. CCS Chem 2023;5:1931-41.
164. Zhang J, Liu X, Ji Y, et al. Atomic-thick metastable phase RhMo nanosheets for hydrogen oxidation catalysis. Nat Commun 2023;14:1761.
165. Yang C, Li Y, Yue J, Cong H, Luo W. Promoting water formation in sulphate-functionalized Ru for efficient hydrogen oxidation reaction under alkaline electrolytes. Chem Sci 2023;14:6289-94.
166. Pi Y, Zhang N, Guo S, Guo J, Huang X. Ultrathin laminar Ir superstructure as highly efficient oxygen evolution electrocatalyst in broad pH range. Nano Lett 2016;16:4424-30.
167. Bu L, Tang C, Shao Q, Zhu X, Huang X. Three-dimensional Pd3Pb nanosheet assemblies: high-performance non-Pt electrocatalysts for bifunctional fuel cell reactions. ACS Catal 2018;8:4569-75.
168. Liu L, Akhoundzadeh H, Li M, Huang H. Alloy catalysts for electrocatalytic CO2 reduction. Small Methods 2023;7:e2300482.
169. Chen H, Zhang B, Liang X, Zou X. Light alloying element-regulated noble metal catalysts for energy-related applications. Chin J Catal 2022;43:611-35.
170. Zhang S, Saji SE, Yin Z, Zhang H, Du Y, Yan CH. Rare-earth incorporated alloy catalysts: synthesis, properties, and applications. Adv Mater 2021;33:e2005988.
171. Yu W, Porosoff MD, Chen JG. Review of Pt-based bimetallic catalysis: from model surfaces to supported catalysts. Chem Rev 2012;112:5780-817.
172. Kitchin JR, Nørskov JK, Barteau MA, Chen JG. Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces. Phys Rev Lett 2004;93:156801.
173. Kandoi S, Ferrin PA, Mavrikakis M. Hydrogen on and in selected overlayer near-surface alloys and the effect of subsurface hydrogen on the reactivity of alloy surfaces. Top Catal 2010;53:384-92.
174. Wang H, Yang Y, Disalvo FJ, Abruña HD. Multifunctional electrocatalysts: Ru–M (M = Co, Ni, Fe) for alkaline fuel cells and electrolyzers. ACS Catal 2020;10:4608-16.
175. Nørskov JK, Abild-Pedersen F, Studt F, Bligaard T. Density functional theory in surface chemistry and catalysis. Proc Natl Acad Sci U S A 2011;108:937-43.
176. Nakaya Y, Furukawa S. Catalysis of alloys: classification, principles, and design for a variety of materials and reactions. Chem Rev 2023;123:5859-947.
177. Ishikawa K, Ohyama J, Okubo K, Murata K, Satsuma A. Enhancement of alkaline hydrogen oxidation reaction of Ru-Ir alloy nanoparticles through bifunctional mechanism on Ru-Ir pair site. ACS Appl Mater Interfaces 2020;12:22771-7.
178. Fu L, Li Y, Yao N, Yang F, Cheng G, Luo W. IrMo nanocatalysts for efficient alkaline hydrogen electrocatalysis. ACS Catal 2020;10:7322-7.
179. Gao X, Wang Y, Xie H, Liu T, Chu W. High activity of a Pt decorated Ni/C nanocatalyst for hydrogen oxidation. Chin J Catal 2017;38:396-403.
180. Zhu S, Qin X, Xiao F, et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat Catal 2021;4:711-8.
181. Luo H, Wang K, Lin F, et al. Amorphous MoOx with high oxophilicity interfaced with PtMo alloy nanoparticles boosts anti-CO hydrogen electrocatalysis. Adv Mater 2023;35:e2211854.
182. Zhang T, Walsh AG, Yu J, Zhang P. Single-atom alloy catalysts: structural analysis, electronic properties and catalytic activities. Chem Soc Rev 2021;50:569-88.
183. Giannakakis G, Flytzani-Stephanopoulos M, Sykes ECH. Single-atom alloys as a reductionist approach to the rational design of heterogeneous catalysts. Acc Chem Res 2019;52:237-47.
184. Gao H, Jiang Y, Chen R, et al. Alloyed Pt single-atom catalysts for durable PEM water electrolyzer. Adv Funct Materials 2023;33:2214795.
185. Mao J, He CT, Pei J, et al. Isolated Ni atoms dispersed on ru nanosheets: high-performance electrocatalysts toward hydrogen oxidation reaction. Nano Lett 2020;20:3442-8.
186. Cai J, Zhang X, Lyu Z, et al. Host-guest ensemble effect on dual-Pt atom-on-Rh nanosheets enables high-efficiency and anti-CO alkaline hydrogen oxidation. ACS Catal 2023;13:6974-82.
187. Zhou M, Li C, Fang J. Noble-metal based random alloy and intermetallic nanocrystals: syntheses and applications. Chem Rev 2021;121:736-95.
188. Zhang B, Fu G, Li Y, et al. General strategy for synthesis of ordered Pt3M intermetallics with ultrasmall particle size. Angew Chem Int Ed Engl 2020;59:7857-63.
189. Zhang J, Shen L, Jiang Y, Sun S. Random alloy and intermetallic nanocatalysts in fuel cell reactions. Nanoscale 2020;12:19557-81.
190. Li Q, Wu L, Wu G, et al. New approach to fully ordered fct-FePt nanoparticles for much enhanced electrocatalysis in acid. Nano Lett 2015;15:2468-73.
191. Yan Y, Du JS, Gilroy KD, Yang D, Xia Y, Zhang H. Intermetallic nanocrystals: syntheses and catalytic applications. Adv Mater 2017:29.
192. Alloyeau D, Ricolleau C, Mottet C, et al. Size and shape effects on the order-disorder phase transition in CoPt nanoparticles. Nat Mater 2009;8:940-6.
193. Luo M, Sun Y, Wang L, Guo S. Tuning multimetallic ordered intermetallic nanocrystals for efficient energy electrocatalysis. Advanced Energy Materials 2017;7:1602073.
194. Zhang J, Zhang L, Cui Z. Strategies to enhance the electrochemical performances of Pt-based intermetallic catalysts. Chem Commun 2021;57:11-26.
195. Kuang P, Ni Z, Zhu B, Lin Y, Yu J. Modulating the d-band center enables ultrafine Pt3 Fe alloy nanoparticles for pH-universal hydrogen evolution reaction. Adv Mater 2023;35:e2303030.
196. Du XX, He Y, Wang XX, Wang JN. Fine-grained and fully ordered intermetallic PtFe catalysts with largely enhanced catalytic activity and durability. Energy Environ Sci 2016;9:2623-32.
197. Lai D, Cheng Q, Zheng Y, et al. A heteronuclear bimetallic organic molecule enabling targeted synthesis of an efficient Pt1Fe1 intermetallic compound for oxygen reduction reaction. J Mater Chem A 2022;10:16639-45.
198. Sun S, Murray CB, Weller D, Folks L, Moser A. Monodisperse FePt nanoparticles and ferromagnetic FePt nanocrystal superlattices. Science 2000;287:1989-92.
199. Wang XX, Hwang S, Pan YT, et al. Ordered Pt3Co intermetallic nanoparticles derived from metal-organic frameworks for oxygen reduction. Nano Lett 2018;18:4163-71.
200. Chung DY, Jun SW, Yoon G, et al. Highly durable and active PtFe nanocatalyst for electrochemical oxygen reduction reaction. J Am Chem Soc 2015;137:15478-85.
201. Jana R, Subbarao U, Peter SC. Ultrafast synthesis of flower-like ordered Pd3Pb nanocrystals with superior electrocatalytic activities towards oxidation of formic acid and ethanol. J Power Sources 2016;301:160-9.
202. Kumar VB, Sanetuntikul J, Ganesan P, Porat Z, Shanmugam S, Gedanken A. Sonochemical formation of Ga-Pt intermetallic nanoparticles embedded in graphene and its potential use as an electrocatalyst. Electrochimica Acta 2016;190:659-67.
203. Heise M, Chang J, Schönemann R, Herrmannsdörfer T, Wosnitza J, Ruck M. Full access to nanoscale bismuth-palladium intermetallics by low-temperature syntheses. Chem Mater 2014;26:5640-6.
204. Cable RE, Schaak RE. Low-temperature solution synthesis of nanocrystalline binary intermetallic compounds using the polyol process. Chem Mater 2005;17:6835-41.
205. Bortoloti F, Garcia A, Angelo A. Electronic effect in intermetallic electrocatalysts with low susceptibility to CO poisoning during hydrogen oxidation. Int J Hydrogen Energy 2015;40:10816-24.
206. Su L, Fan X, Jin Y, Cong H, Luo W. Hydroxyl-binding energy-induced kinetic gap narrowing between acidic and alkaline hydrogen oxidation reaction on intermetallic Ru3Sn7 catalyst. Small 2023;19:e2207603.
207. Zhao Y, Yang F, Zhang W, et al. High-performance Ru2P anodic catalyst for alkaline polymer electrolyte fuel cells. CCS Chem 2022;4:1732-44.
208. Su L, Jin Y, Fan X, Liu Z, Luo W. pH-Dependent binding energy-induced inflection-point behaviors for pH-universal hydrogen oxidation reaction. Sci China Chem 2023;66:3262-8.
209. Huang H, Liu K, Yang F, et al. Breaking surface atomic monogeneity of rh2p nanocatalysts by defect-derived phosphorus vacancies for efficient alkaline hydrogen oxidation. Angew Chem Int Ed Engl 2023;62:e202315752.
210. Su L, Chen J, Yang F, et al. Electric-double-layer origin of the kinetic ph effect of hydrogen electrocatalysis revealed by a universal hydroxide adsorption-dependent inflection-point behavior. J Am Chem Soc 2023;145:12051-8.
211. Deng Y, Tripkovic V, Rossmeisl J, Arenz M. Oxygen reduction reaction on Pt overlayers deposited onto a gold film: ligand, strain, and ensemble effect. ACS Catal 2016;6:671-6.
212. Zhao X, Sasaki K. Advanced Pt-based core-shell electrocatalysts for fuel cell cathodes. Acc Chem Res 2022;55:1226-36.
213. Jiang R, Tung SO, Tang Z, et al. A review of core-shell nanostructured electrocatalysts for oxygen reduction reaction. Energy Storage Mater 2018;12:260-76.
214. Hammer B, Nørskov J. Theoretical surface science and catalysis-calculations and concepts. Impact of Surface Science on Catalysis. Elsevier; 2000. pp. 71-129.
215. Gong T, Alghamdi H, Raciti D, Hall AS. Improved alkaline hydrogen oxidation on strain-modulated Pt overlayers at ordered intermetallic Pt–Sb cores. ACS Energy Lett 2023;8:685-90.
216. Schwämmlein JN, Stühmeier BM, Wagenbauer K, et al. Origin of superior HOR/HER activity of bimetallic Pt-Ru catalysts in alkaline media identified via Ru@Pt core-shell nanoparticles. J Electrochem Soc 2018;165:H229-39.
217. Cai J, Liao X, Li P, et al. Penta-twinned Rh@Pt core-shell nanobranches with engineered shell thickness for reversible and active hydrogen redox electrocatalysis. Chem Eng J 2022;429:132414.
218. Du H, Du Z, Wang T, et al. Unlocking interfacial electron transfer of ruthenium phosphides by homologous core-shell design toward efficient hydrogen evolution and oxidation. Adv Mater 2022;34:e2204624.
219. Luo Z, Zhao G, Pan H, Sun W. Strong metal–support interaction in heterogeneous catalysts. Adv Energy Mater 2022;12:2201395.
220. Sun H, Tung CW, Qiu Y, et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J Am Chem Soc 2022;144:1174-86.
221. Chen J, Zhang Y, Zhang Z, et al. Metal–support interactions for heterogeneous catalysis: mechanisms, characterization techniques and applications. J Mater Chem A 2023;11:8540-72.
222. Kundu MK, Mishra R, Bhowmik T, Barman S. Rhodium metal–rhodium oxide (Rh-Rh2O3) nanostructures with Pt-like or better activity towards hydrogen evolution and oxidation reactions (HER, HOR) in acid and base: correlating its HOR/HER activity with hydrogen binding energy and oxophilicity of the catalyst. J Mater Chem A 2018;6:23531-41.
223. Miller HA, Lavacchi A, Vizza F, et al. A Pd/C-CeO2 anode catalyst for high-performance platinum-free anion exchange membrane fuel cells. Angew Chem Int Ed Engl 2016;55:6004-7.
224. Singh RK, Davydova ES, Douglin J, et al. Synthesis of CeOx -decorated Pd/C catalysts by controlled surface reactions for hydrogen oxidation in anion exchange membrane fuel cells. Adv Funct Materials 2020;30:2002087.
225. Bhowmik T, Kundu MK, Barman S. Palladium nanoparticle–graphitic carbon nitride porous synergistic catalyst for hydrogen evolution/oxidation reactions over a broad range of pH and correlation of its catalytic activity with measured hydrogen binding energy. ACS Catal 2016;6:1929-41.
226. Zhou Y, Xie Z, Jiang J, et al. Lattice-confined Ru clusters with high CO tolerance and activity for the hydrogen oxidation reaction. Nat Catal 2020;3:454-62.
227. Li Z, Ji S, Liu Y, et al. Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites. Chem Rev 2020;120:623-82.
228. Zhu Y, Zhang X, Koh K, et al. Inverse iron oxide/metal catalysts from galvanic replacement. Nat Commun 2020;11:3269.
229. Rodriguez JA, Liu P, Graciani J, et al. Inverse oxide/metal catalysts in fundamental studies and practical applications: a perspective of recent developments. J Phys Chem Lett 2016;7:2627-39.
230. Chen G, Zhao Y, Fu G, et al. Interfacial effects in iron-nickel hydroxide-platinum nanoparticles enhance catalytic oxidation. Science 2014;344:495-9.
231. Fu Q, Yang F, Bao X. Interface-confined oxide nanostructures for catalytic oxidation reactions. Acc Chem Res 2013;46:1692-701.
232. Lyu Z, Zhang XG, Wang Y, et al. Amplified interfacial effect in an atomically dispersed RuOx-on-Pd 2D inverse nanocatalyst for high-performance oxygen reduction. Angew Chem Int Ed Engl 2021;60:16093-100.
233. Fu L, Yang F, Hu Y, Li Y, Chen S, Luo W. Discrepant roles of adsorbed OH* species on IrWOx for boosting alkaline hydrogen electrocatalysis. Sci Bull 2020;65:1735-42.
234. Ma M, Li G, Yan W, et al. Single-atom molybdenum engineered platinum nanocatalyst for boosted alkaline hydrogen oxidation. Adv Energy Mater 2022;12:2103336.
235. Huang Z, Lu R, Zhang Y, et al. A highly efficient pH-universal HOR catalyst with engineered electronic structures of single Pt sites by isolated Co atoms. Adv Funct Materials 2023;33:2306333.
236. Zhou F, Ke X, Chen Y, et al. Electron-distribution control via Pt/NC and MoC/NC dual junction: boosted hydrogen electro-oxidation and theoretical study. J Energy Chem 2024;88:513-20.
237. Zhang Y, Li G, Zhao Z, et al. Atomically isolated Rh sites within highly branched Rh2Sb nanostructures enhance bifunctional hydrogen electrocatalysis. Adv Mater 2021;33:e2105049.
238. Wang H, Abruña HD. Rh and Rh alloy nanoparticles as highly active H2 oxidation catalysts for alkaline fuel cells. ACS Catal 2019;9:5057-62.
239. Su L, Zhao Y, Jin Y, Fan X, Liu Z, Luo W. d–p Orbital hybridization in RhSn catalyst boosts hydrogen oxidation reaction under alkaline electrolyte. J Mater Chem A 2022;10:21856-61.
240. Ming M, Zhang Y, He C, et al. Room-temperature sustainable synthesis of selected platinum group metal (PGM = Ir, Rh, and Ru) nanocatalysts well-dispersed on porous carbon for efficient hydrogen evolution and oxidation. Small 2019;15:e1903057.
241. Su L, Zhao Y, Yang F, Wu T, Cheng G, Luo W. Ultrafine phosphorus-doped rhodium for enhanced hydrogen electrocatalysis in alkaline electrolytes. J Mater Chem A 2020;8:11923-7.
242. Zhao Y, Wang X, Cheng G, Luo W. Phosphorus-induced activation of ruthenium for boosting hydrogen oxidation and evolution electrocatalysis. ACS Catal 2020;10:11751-7.
243. Jiang J, Tao S, He Q, et al. Interphase-oxidized ruthenium metal with half-filled d-orbitals for hydrogen oxidation in an alkaline solution. J Mater Chem A 2020;8:10168-74.
244. Zhao Y, Wu D, Luo W. Correlating alkaline hydrogen electrocatalysis and hydroxide binding energies on Mo-modified Ru catalysts. ACS Sustainable Chem Eng 2022;10:1616-23.
245. Wang P, Wang C, Yang Y, et al. RuP nanoparticles supported on N, O codoped porous hollow carbon for efficient hydrogen oxidation reaction. Adv Materials Inter 2022;9:2102193.
246. Wang J, Liu J, Zhang B, et al. Amine-ligand modulated ruthenium nanoclusters as a superior bi-functional hydrogen electrocatalyst in alkaline media. J Mater Chem A 2021;9:22934-42.
247. Xue Y, Shi L, Liu X, et al. A highly-active, stable and low-cost platinum-free anode catalyst based on RuNi for hydroxide exchange membrane fuel cells. Nat Commun 2020;11:5651.
248. Liu D, Lu S, Xue Y, et al. One-pot synthesis of IrNi@Ir core-shell nanoparticles as highly active hydrogen oxidation reaction electrocatalyst in alkaline electrolyte. Nano Energy 2019;59:26-32.
249. Ji X, Chen P, Liu Y, et al. Ir/Ni–NiO/CNT composites as effective electrocatalysts for hydrogen oxidation. J Mater Chem A 2023;11:5076-82.
250. Liu D, Luo Z, Zhang B, et al. Tailoring interfacial charge transfer of epitaxially grown Ir clusters for boosting hydrogen oxidation reaction. Adv Energy Mater 2023;13:2202913.
251. Su L, Zhao Y, Jin Y, Liu Z, Cui H, Luo W. Identifying the role of hydroxyl binding energy in a non-monotonous behavior of Pd-Pd4S for hydrogen oxidation reaction. Adv Funct Materials 2022;32:2113047.
252. Qin B, Yu H, Jia J, et al. A novel IrNi@PdIr/C core-shell electrocatalyst with enhanced activity and durability for the hydrogen oxidation reaction in alkaline anion exchange membrane fuel cells. Nanoscale 2018;10:4872-81.
253. Qiu Y, Xin L, Li Y, et al. BCC-phased PdCu alloy as a highly active electrocatalyst for hydrogen oxidation in alkaline electrolytes. J Am Chem Soc 2018;140:16580-8.
254. Pang B, Jia C, Wang S, et al. Self-optimized ligand effect of single-atom modifier in ternary pt-based alloy for efficient hydrogen oxidation. Nano Lett 2023;23:3826-34.
255. Hamo ER, Singh RK, Douglin JC, et al. Carbide-supported PtRu catalysts for hydrogen oxidation reaction in alkaline electrolyte. ACS Catal 2021;11:932-47.
256. Zhang J, Qu X, Shen L, et al. Engineering the near-surface of PtRu3 nanoparticles to improve hydrogen oxidation activity in alkaline electrolyte. Small 2021;17:e2006698.
257. Jin Y, Chen F, Wang J, Guo L, Jin T, Liu H. Lamellar platinum-rhodium aerogels with superior electrocatalytic performance for both hydrogen oxidation and evolution reaction in alkaline environment. J Power Sources 2019;435:226798.
258. Zhan C, Xu Y, Bu L, et al. Subnanometer high-entropy alloy nanowires enable remarkable hydrogen oxidation catalysis. Nat Commun 2021;12:6261.
259. Pan Z, An L, Zhao T, Tang Z. Advances and challenges in alkaline anion exchange membrane fuel cells. Progress in Energy and Combustion Science 2018;66:141-75.
260. Pinaud BA, Bonakdarpour A, Daniel L, Sharman J, Wilkinson DP. Key considerations for high current fuel cell catalyst testing in an electrochemical half-cell. J Electrochem Soc 2017;164:F321-7.
261. Chung HT, Martinez U, Matanovic I, Kim YS. Cation-hydroxide-water coadsorption inhibits the alkaline hydrogen oxidation reaction. J Phys Chem Lett 2016;7:4464-9.
262. Maurya S, Fujimoto CH, Hibbs MR, Narvaez Villarrubia C, Kim YS. Toward improved alkaline membrane fuel cell performance using quaternized aryl-ether free polyaromatics. Chem Mater 2018;30:2188-92.
263. Maurya S, Dumont JH, Villarrubia CN, et al. Surface adsorption affects the performance of alkaline anion-exchange membrane fuel cells. ACS Catal 2018;8:9429-39.
264. Yassin K, Douglin JC, Rasin IG, et al. The effect of membrane thickness on AEMFC Performance: an integrated theoretical and experimental study. Energy Convers Manage 2022;270:116203.
265. Dekel DR. Unraveling mysteries of hydrogen electrooxidation in anion exchange membrane fuel cells. Curr Opin Electrochem 2018;12:182-8.
266. León MI, Valentín-reyes J, Romero-castañón T, Beltrán J, Flores-hernández JR, Nava JL. Water movement through an anion exchange membrane fuel cell (AEMFC): Influence of gas humidity and flow rate. Appl Energy 2022;324:119722.
267. Ni W, Wang T, Schouwink PA, Chuang YC, Chen HM, Hu X. Efficient hydrogen oxidation catalyzed by strain-engineered nickel nanoparticles. Angew Chem Int Ed Engl 2020;59:10797-801.
268. Alesker M, Page M, Shviro M, et al. Palladium/nickel bifunctional electrocatalyst for hydrogen oxidation reaction in alkaline membrane fuel cell. J Power Sources 2016;304:332-9.
269. Omasta TJ, Peng X, Miller HA, et al. Beyond 1.0 W cm−2 performance without platinum: the beginning of a new era in anion exchange membrane fuel cells. J Electrochem Soc 2018;165:J3039-44.
270. Miller HA, Vizza F, Marelli M, et al. Highly active nanostructured palladium-ceria electrocatalysts for the hydrogen oxidation reaction in alkaline medium. Nano Energy 2017;33:293-305.
271. Wang Y, Wang G, Li G, et al. Pt–Ru catalyzed hydrogen oxidation in alkaline media: oxophilic effect or electronic effect? Energy Environ Sci 2015;8:177-81.
272. Li Q, Peng H, Wang Y, Xiao L, Lu J, Zhuang L. The comparability of Pt to Pt-Ru in catalyzing the hydrogen oxidation reaction for alkaline polymer electrolyte fuel cells operated at 80 °C. Angew Chem Int Ed Engl 2019;58:1442-6.
273. Cong Y, Chai C, Zhao X, Yi B, Song Y. Pt 0.25 Ru 0.75 /N-C as highly active and durable electrocatalysts toward alkaline hydrogen oxidation reaction. Adv Materials Inter 2020;7:2000310.
274. Ni W, Meibom JL, Hassan NU, et al. Synergistic interactions between PtRu catalyst and nitrogen-doped carbon support boost hydrogen oxidation. Nat Catal 2023;6:773-83.
275. Wang R, Li D, Maurya S, et al. Ultrafine Pt cluster and RuO2 heterojunction anode catalysts designed for ultra-low Pt-loading anion exchange membrane fuel cells. Nanoscale Horiz 2020;5:316-24.
276. Zhao T, Li M, Xiao D, et al. Pseudo-Pt monolayer for robust hydrogen oxidation. J Am Chem Soc 2023;145:4088-97.
277. Qin B, Yu H, Gao X, et al. Ultrathin IrRu nanowire networks with high performance and durability for the hydrogen oxidation reaction in alkaline anion exchange membrane fuel cells. J Mater Chem A 2018;6:20374-82.
278. Miller HA, Pagliaro MV, Bellini M, et al. Integration of a Pd-CeO2 /C anode with Pt and Pt-free cathode catalysts in high power density anion exchange membrane fuel cells. ACS Appl Energy Mater 2020;3:10209-14.
279. Ogada JJ, Ipadeola AK, Mwonga PV, et al. CeO2 modulates the electronic states of a palladium onion-like carbon interface into a highly active and durable electrocatalyst for hydrogen oxidation in anion-exchange-membrane fuel cells. ACS Catal 2022;12:7014-29.
280. Ohyama J, Sato T, Satsuma A. High performance of Ru nanoparticles supported on carbon for anode electrocatalyst of alkaline anion exchange membrane fuel cell. J Power Sources 2013;225:311-5.
281. Zeng L, Peng H, Liu W, et al. Extraordinary activity of mesoporous carbon supported Ru toward the hydrogen oxidation reaction in alkaline media. J Power Sources 2020;461:228147.
282. Tatus-Portnoy Z, Kitayev A, Vineesh TV, Tal-Gutelmacher E, Page M, Zitoun D. A low-loading Ru-rich anode catalyst for high-power anion exchange membrane fuel cells. Chem Commun 2020;56:5669-72.
283. Yang C, Li Y, Ge C, et al. The role of hydroxide binding energy in alkaline hydrogen oxidation reaction kinetics on RuCr nanosheet†. Chin J Chem 2022;40:2495-501.
284. Li Y, Yang C, Ge C, et al. Electronic modulation of Ru nanosheet by d-d orbital coupling for enhanced hydrogen oxidation reaction in alkaline electrolytes. Small 2022;18:e2202404.
285. Han P, Yang X, Wu L, et al. A highly-efficient boron interstitially inserted Ru anode catalyst for anion exchange membrane fuel cells. Adv Mater 2023:e2304496.
286. Peng X, Kulkarni D, Huang Y, et al. Using operando techniques to understand and design high performance and stable alkaline membrane fuel cells. Nat Commun 2020;11:3561.
287. Lin XM, Wang XT, Deng YL, et al. In situ probe of the hydrogen oxidation reaction intermediates on PtRu a bimetallic catalyst surface by core-shell nanoparticle-enhanced raman spectroscopy. Nano Lett 2022;22:5544-52.
288. Song F, Li W, Yang J, et al. Interfacial sites between cobalt nitride and cobalt act as bifunctional catalysts for hydrogen electrochemistry. ACS Energy Lett 2019;4:1594-601.
289. Xiong B, Zhao W, Chen L, Shi J. One-step synthesis of W2C@N,P-C nanocatalysts for efficient hydrogen electrooxidation across the whole pH range. Adv Funct Materials 2019;29:1902505.
290. Feng M, Huang J, Peng Y, Huang C, Yue X, Huang S. Tuning the electronic structures of cobalt-molybdenum bimetallic carbides to boost the hydrogen oxidation reaction in alkaline medium. Chem Eng J 2022;428:131206.
291. Kabir S, Lemire K, Artyushkova K, et al. Platinum group metal-free NiMo hydrogen oxidation catalysts: high performance and durability in alkaline exchange membrane fuel cells. J Mater Chem A 2017;5:24433-43.
292. Ni W, Wang T, Héroguel F, et al. An efficient nickel hydrogen oxidation catalyst for hydroxide exchange membrane fuel cells. Nat Mater 2022;21:804-10.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Fu L, Wang S, Cai J, Huang H, Yang F, Xie S. Recent advances in platinum-group-metal based electrocatalysts for alkaline hydrogen oxidation reaction. Chem Synth 2024;4:8. http://dx.doi.org/10.20517/cs.2023.53
AMA Style
Fu L, Wang S, Cai J, Huang H, Yang F, Xie S. Recent advances in platinum-group-metal based electrocatalysts for alkaline hydrogen oxidation reaction. Chemical Synthesis. 2024; 4(2): 8. http://dx.doi.org/10.20517/cs.2023.53
Chicago/Turabian Style
Fu, Luhong, Shupeng Wang, Junlin Cai, Hongpu Huang, Fulin Yang, Shuifen Xie. 2024. "Recent advances in platinum-group-metal based electrocatalysts for alkaline hydrogen oxidation reaction" Chemical Synthesis. 4, no.2: 8. http://dx.doi.org/10.20517/cs.2023.53
ACS Style
Fu, L.; Wang S.; Cai J.; Huang H.; Yang F.; Xie S. Recent advances in platinum-group-metal based electrocatalysts for alkaline hydrogen oxidation reaction. Chem. Synth. 2024, 4, 8. http://dx.doi.org/10.20517/cs.2023.53
About This Article
Special Issue
Copyright

Author Biographies






Data & Comments
Data


 Cite This Article 17 clicks
Cite This Article 17 clicks

 Like This Article 17
likes
Like This Article 17
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.