
Schizophrenic behavior of 2,3-oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs

Abstract
We report the unusual behavior of oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs bearing two alkyl groups different from a methyl, at their Δ18-19 double bond: unambiguous structure determinations of the products and tentative rational for their formation are described.
Keywords
INTRODUCTION
The transformation of linear squalene to (20R)-lanosterol (R)-3a possessing four cycles and seven chiral centers, by membrane-bound oxidosqualene sterol cyclase (OSC; lanosterol synthetase), has been the subject of constant interest for more than 70 years[1-5]. Woodward and Bloch[6] first proposed that lanosterol is an intermediate in the transformation of squalene to cholesterol, and soon after Stork[7,8] and the Zurich School[9,10] independently proposed a detailed mechanism for the construction of the steroid scaffold [Scheme 1].
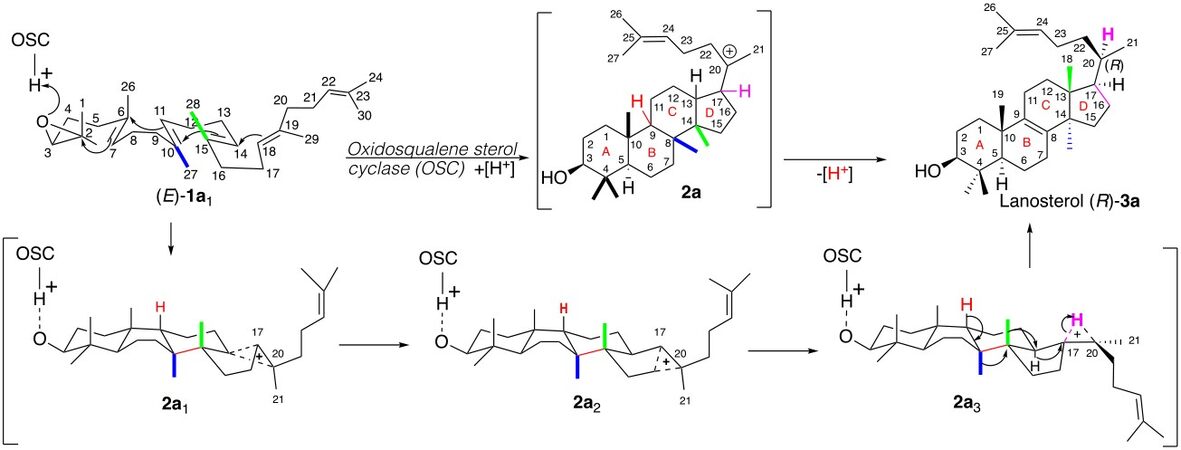
Scheme 1. Description of the Zurich School mechanism from all (E)2,3-oxidosqualenes to lanosterol.
The original description of this mechanism [Scheme 1], adapted to the discovery by Corey et al.[11] and Van Tamelen et al.[12] that 2,3-oxidosqualene (E)-1a is intermediate in this transformation, involves: (i) a polycyclization, implying its Δ6-7, Δ9-10, Δ13-14, and Δ18-19 double bonds, initiated by an acid catalyst part of the enzyme and prefolded by the enzyme in a chair-boat-chair-boat conformation; (ii) this leads to the formation of the elusive intermediate 2a named “protosterol”, possessing a 6,6,6,5-membered polycyclic scaffold (A/B/C/D) and bearing a tertiary carbocation at C-20 (steroid nomenclature) on the side chain lying in α-position; and (iii) a series of 1,2-shifts on the latter, ending by the loss of the (H9) proton delivering lanosterol (R)-3a possessing a Δ8-9 double bond (steroid nomenclature, Scheme 1).
The proposed mechanism is based on two stereochemical assumptions supported by strong experimental evidence: (i) antiperiplanar addition across each C=C double bond; and (ii) suprafacial 1,2-shifts driving each migration (H17, H13, Me14, and Me8).
However, this proposal leads to a protosterol with an alpha-oriented side chain whose stereochemistry at C-20 formerly requires a 120° clockwise rotation around the C17-C20 bond to initiate the series of migrations leading to the lanosterol possessing the natural (20R)-stereochemistry. This is sketched[9,10] in the original proposal by a series of non-classical ion rearrangements (2a1 → 2a2 → 2a3, Scheme 1).
It was then rationalized by Cornforth[13], who hypothesized that the protosterol should resemble 2a4 bearing an exogeneous X group at C-20 on the side chain lying in α-position. It results from: (i) the folding of the polyene originally in a chair-boat-chair-boat conformation (E)-1a1 as previously reported by the Zurich School (Scheme 2, entry a); (ii) a series of concerted anti-addition of the different [C=C] double bonds ending by the addition on the [Cb=Cc] double bond of [Ca] (from the re face) on [Cb] and an exogeneous X group at [Cc] (from the re face) that produces concomitantly the D ring; and (iii) a 120° clockwise rotation of the side chain, located in α-position, around the [Cb-Cc] single bond leading to 2a4, in which the Cb-X bond is aligned with the Cb-H bond in an antiperiplanar conformation that initiates the departure of the X group and the series of migrations leading to the (20R)-lanosterol (20R)-3a (Scheme 2, entry a).
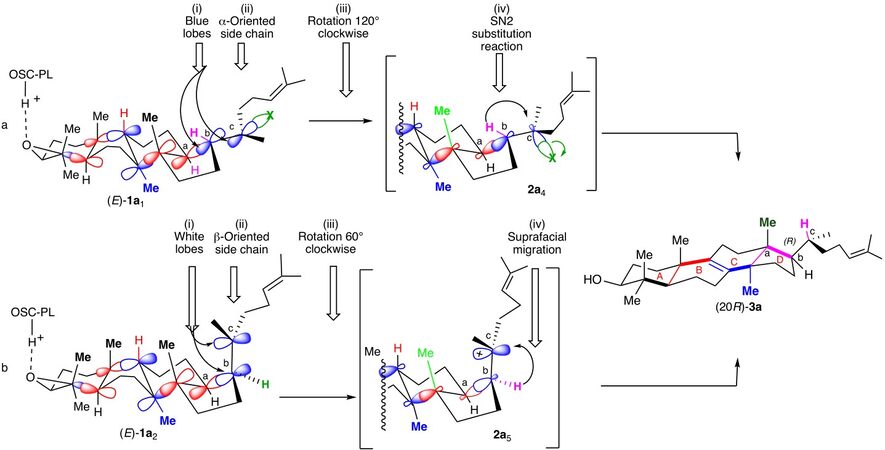
Scheme 2. Cornforth (entry a) and Corey (entry b) mechanisms accounting for the (R)-stereochemistry of lanosterol 3a at C-20.
The Corey proposal (Scheme 2, entry b)[14,15] avoids the 120° rotation implied in each of the previous mechanisms by involving the intermediate formation of a protosterol 2a5 resulting from a chair-boat-chair-chair folding of the 2,3-oxidopolyene (E)-1a2, different from the above, and bearing its side chain in β-position. It accordingly results from the attack of Ca by the si face of Cb (Scheme 2, entry b). A rotation of the chain of only 60° clockwise around the Cb-Cc allows the alignment of the Cb-H bond with the π-bond of the carbocation at Cc and initiates the series of migrations leading to the (20R)-lanosterol (20R)-3a (Scheme 2, entry b)[1-5,14,15].
The latter mechanism, experimentally supported[14,15] by isolation of a stable protosterol analog in which the carbocation at C-20 is trapped by water from a suitable oxidosqualene analog, is now generally admitted[1-5].
Since the polycyclization process ends and the series of migration starts at the C-20 carbon, we expected to obtain precious information on the whole process by looking at what happens there. We therefore initiated, a long ago, a research program[16-18] aimed at gathering the behavior of oxidopolyenes analogs that differ from the original by the nature of the substituents at C-19 and the stereochemistry of their Δ18-19 double bond towards oxidosqualene sterol cyclases extracted from mammals (pig liver, OSC-PL) or yeast
RESULTS AND DISCUSSION
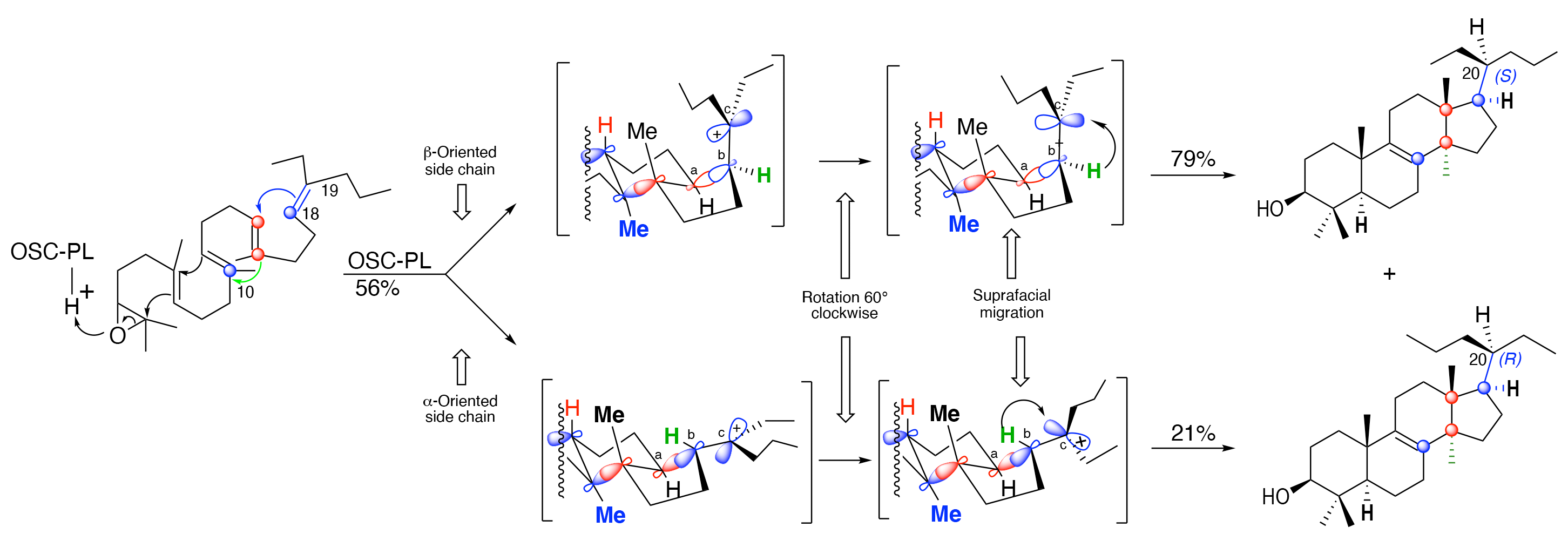
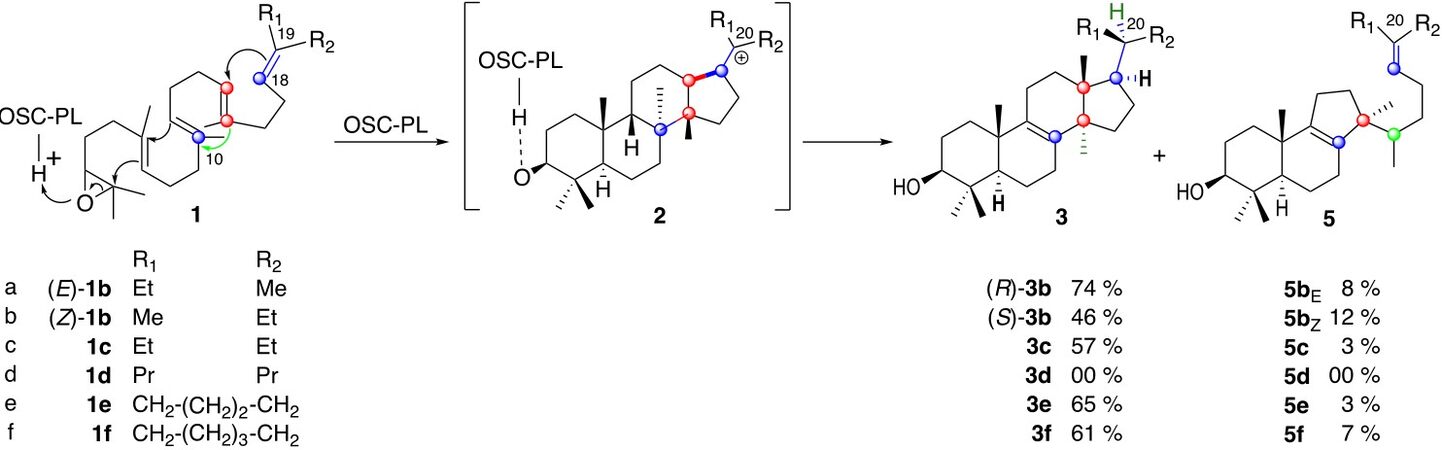
We report the first example of a transformation of an oxidosqualene analog by OSC-PL that leads to a mixture of lanosterol analog epimers at C-20 whose formation relates to carbocationic protosterols in which the side chain lies for the major one in the β-position, as disclosed in the Corey mechanism (Scheme 2, entry b), and for the minor one in the α-position, as originally proposed in the Zurich-Stork mechanism (detailed in part in Scheme 2, entry a). These results contrast with previous results from our laboratory involving related oxidosqualene analogs that exhibit complete stereocontrol.
We previously reported that oxidosqualene analogs (E)-1b and (Z)-1b possessing a methyl and an ethyl group at C-19 with the natural (E)- and the unnatural (Z)-stereochemistry at their Δ18-19 double bond are mainly cyclized by OSC-PL to lanosterol analogs 3b that only differ from their stereochemistry at C-20, along with a few percent of the tricyclic compounds 5b resulting from a partial polycyclization (Scheme 3, compare entries a and b)[16,17].
Scheme 3. Results concerning the transformation of truncated 2,3-oxidosqualene analogs by OSC-PL.
However, the reaction of OSC-PL[19] with the 2,3-oxidosqualene analog (Z)-1a possessing the same hydrocarbon framework as the natural product (E)-1a but inverted stereochemistry at C-19 does not produce the 6.6.6.5 tetracyclic lanosterol epimer at C-20 but delivers the tricyclic 6.6.5 derivatives (Z)-6a and (Z)-7a. It results from partial polycyclizations that do not involve the participation of the Δ18-19 double bond
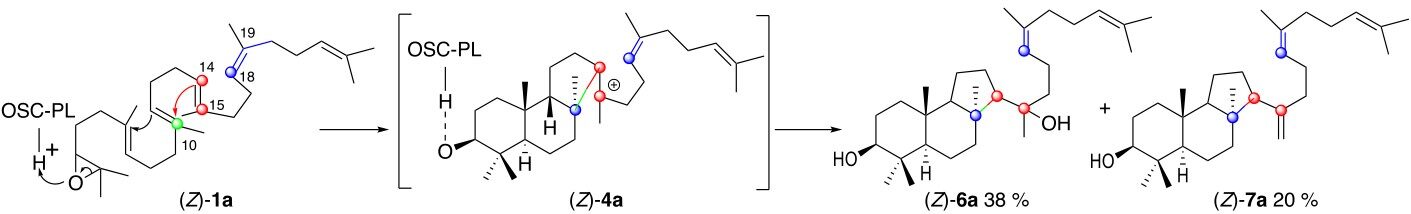
Scheme 4. Results concerning the transformation of a stereoisomeric 2,3-oxidosqualene by OSC-PL.
Since then, a few other oxidopolyenes have been reacted with OSC-PL, and it was observed that the amount of tetracyclic compounds over the tricyclic ones decreases dramatically by increasing the length of the side chain attached at C-19 in the Δ18-19 unnatural (Z)-series, whereas the reverse was found in the (E)-series[17,21].
We then became aware that changing the stereochemistry of the oxidopolyene has a dual impact on its interactions with the enzyme: It increases the interaction on the one side (trans to the hydrogen at C-18), but, at the same time, it decreases the interactions with the other side (cis to the hydrogen at C-18). We therefore decided to study the behavior, towards pig liver OSC-PL[19], of oxidosqualene analog 1 bearing at C-19 two alkyl groups different from a methyl group.
We now report that the 2,3-oxidosqualene analog 1c bearing two ethyl at C-19 mainly produces the lanosterol analog 1c bearing two ethyl groups at C-20 (Scheme 3, entry c), whereas its higher homolog 1d bearing two propyl groups there is recovered unchanged under similar or even more drastic conditions (3 or 7 h reaction at 22 °C, Scheme 3, entry d), suggesting that it is not accepted by the enzyme. Interestingly, constraining the mobility of those two chains by incorporating them into a five-membered ring as in 1e or a six-membered cycle as in 1f is not deleterious for the formation of the lanosterol analogs 3e and 3f (Scheme 3, entries e and f).
Testing the behavior of 2,3-oxidosqualene analog 1g bearing at C-19 an ethyl and a propyl group towards pig liver OSC-PL was obviously our next objective, hoping that at least one of the two stereoisomers at Δ18-19 would cyclize.
We laboriously prepared each of the two stereoisomers (E)-1g and (Z)-1g in pure form and unexpectedly found that each of them on incubation with pig liver OSC-PL delivers, in fair yields, lanosterol analog 3g possessing a propyl and an ethyl group at C-20 [Scheme 5]. Careful investigations and comparison of the biosynthetic compounds with authentic sample of (20S)-3g and (20R)-3g synthesized independently, as reported below, unambiguously shows that the oxidosqualene (E)-1g possessing a E-Δ18-19 C,C double bond produces the lanosterol analog (R)-3g possessing the (20R)-stereochemistry in 43% yield
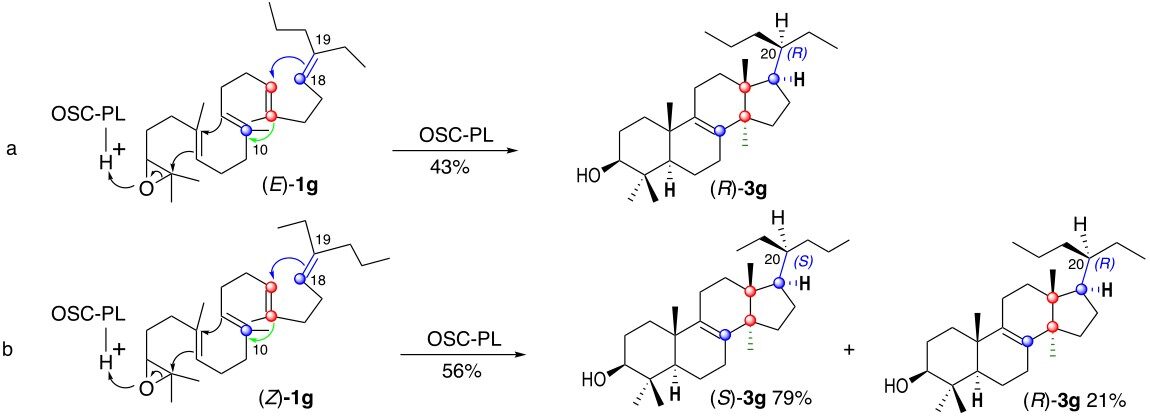
Scheme 5. OSC-PL behavior towards 2,3-oxidosqualene analogs bearing (E)- and (Z)-stereochemistry at Δ18-19.
The results in Schemes 3-5 clearly show the importance of the length of the chains attached at C-19 on 2,3-oxidosqualene analogs towards OSC-PL. Comparing the results reported in Scheme 5 to those disclosed in Scheme 3, entry d, suggests the exceptional role of an “added carbon” in preventing the quite far removed oxido moiety from reaching the enzymic active site. Comparison of the behavior of 1g to that of 1b (Schemes 3-5, entries a and b) leads to a change in the status of the process from stereospecific to stereoselective.
The Corey model (Scheme 2, entry b)[14,15] fits well to rationalize the formation of (20R)-3g from the oxidosqualene analog (E)-1g (Scheme 6, entry a) as well as that of the major diastereoisomer of the sterol (20S)-3g obtained from the oxidosqualene analog (Z)-1g.
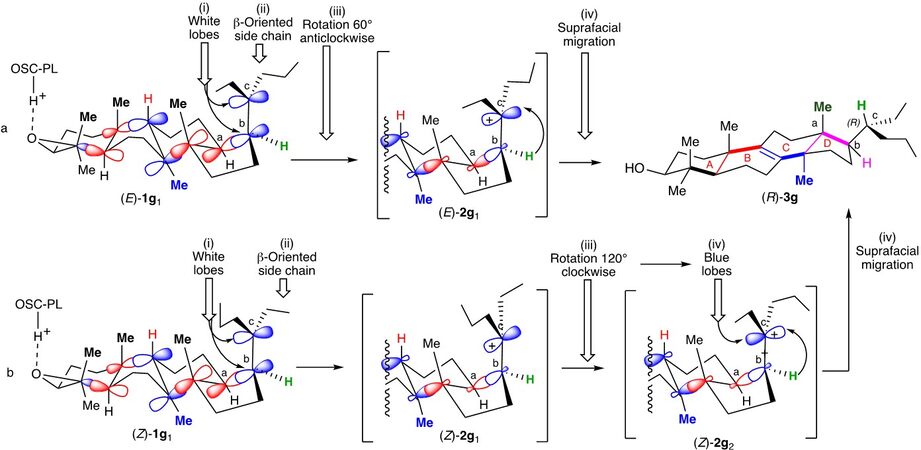
Scheme 6. Rationalizing with the use of the Corey mechanism the formation of (R)-3g from (E)-1g and (Z)-1g.
It is however less obvious to apply the Corey model to the formation of the minor isomer (20R)-3g from (Z)-1g (Scheme 6, entry b) since it requires an anticlockwise rotation of 120° of the side chain around the
A related 120° rotation around the C17-C20 bond in the model disclosed in Scheme 2, entry a, was previously rejected by Corey et al.[14] and is at the origin of its mechanistic model (Scheme 2, entry b)[14,15]. It is interesting to note that, although the Corey model could eventually be adapted, as discussed above, the Cornforth model[13] (Scheme 2, entry a) could not be applied to explain this experimental result.
Could it be that the presence of the 3-isohexenyl group in such stereochemical arrangement favors, at least partly, a chair-boat-chair-boat conformation involving the (Z)-1g2 intermediate [Scheme 7] reminiscent of the original report of the Zurich School [Scheme 1][9,10] over the chair-boat-chair-chair conformation involving the (Z)-1g1 intermediate required by the Corey model [Scheme 6][14,15]?
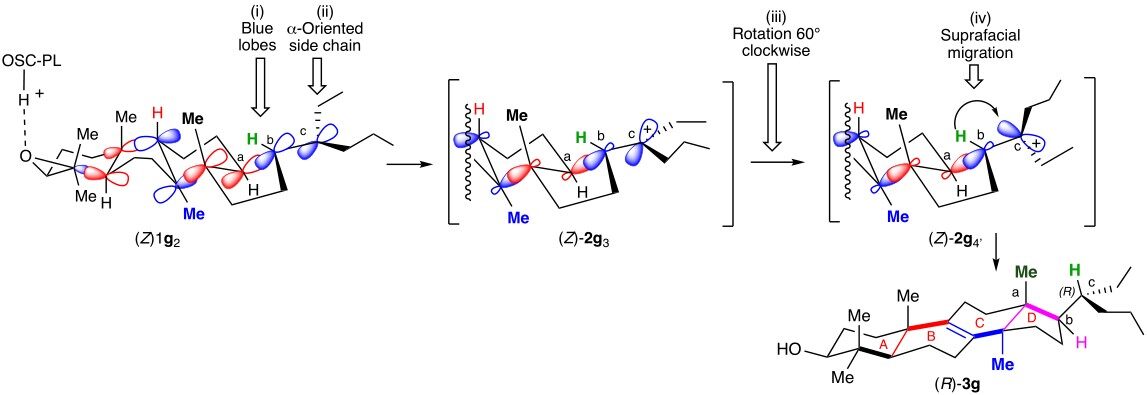
Scheme 7. Rationalizing with the use of the adapted Zurich School mechanism the formation of (R)-3g from (Z)-1g.
In such case, the whole sequence of antiperiplanar additions on (Z)-1g2would lead to the protosterol
The series of results reported here seems to allow for some fine tuning regarding the interactions between the oxidosqualenes analog 1 and the enzymic environment of OSC-PL. We should refrain from the use of generalizations to deduce the intimate mechanism of lanosterol biosynthesis derived from specific experiments as it has been very often done.
Apparently, OSC-PL possesses an exceptional propensity to adapt its behavior to the substrate, a behavior it does not share for example with its distant ancestor cyclase, OSC-SC, which does not accept at all the Δ17-18 (Z)-analogs of 2,3-oxidosqualene tested[14,17].
EXPERIMENTAL
This study was carried out on C-14 radiolabeled racemic mixtures of 2,3-oxidopolyene 1 (epimeric at C-3) bearing either (E)- or (Z)-stereochemistry at Δ18-19 and therefore the yields were adjusted to the fact that only one enantiomer, the one possessing the (S)-stereochemistry at C-3, is biotransformed. The syntheses reported below and “classical” purifications provided at best samples of 1g with a content of 10% of the other diastereoisomer. Isolation of each pure stereoisomer from each 1g mixture (HPLC; Hypercarb
Synthesis of 2,3-oxidopolyenes
The synthesis of the 2,3-oxidopolyenes 1d-f was achieved [Scheme 8] from the 2,3-oxidopolyenic aldehyde[17,23] 8
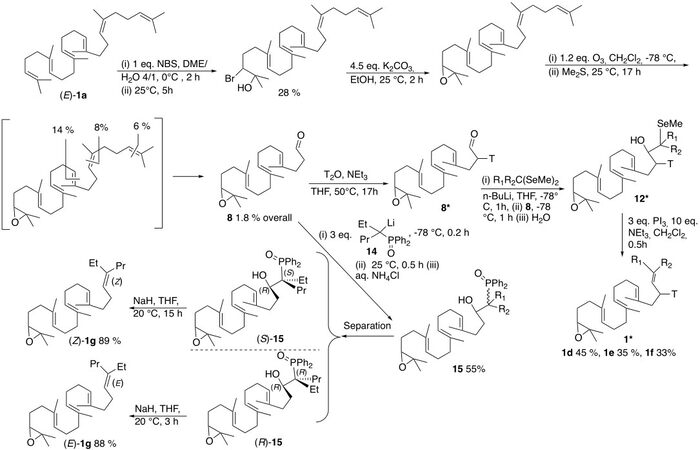
Scheme 8. Description of the synthetic schemes allowing the synthesis of 2,3-oxidosqualene analogs bearing different substituents and stereochemistry at Δ18-19.
The 2,3-oxidopolyenic aldehyde 8 has been prepared (28% yield) from squalene and N-bromosuccinimide in H2O/DME[23], followed by treatment with excess K2CO3 (4.5 eq.) in methanol and ozonolysis of the resulting epoxide (88% yield) with ozone [(i) 1.2 eq. CH2Cl2, (ii) excess Me2S; yield in 8: 8%].
The synthesis of 1g was performed using Horner-Warren method[26,27] since we were unable to separate the related stereoisomeric mixture of β-hydroxyalkylselenide 12g [Scheme 8]. Thus, the 2,3-oxidopolyenic aldehyde 8 was reacted with an excess of ylide 14 generated from 3-diphenylphosphinyloxyhexane 13 [(i) 3 eq. 13, 6 eq. LDA, THF, 0 °C 0.2 h; (ii) -78 °C; (iii) 1eq. 8, THF, -78 °C, 0.2 h; (iv) 20 °C, 0.5 h; and (v) aq. NH4Cl], leading to a stereoisomeric mixture of β-hydroxyalkylphosphinyloxide 15 (55% yield), separated on a silica gel column (ether/pentane of 9/1, as two 90/10 mixtures after several subsequent separations). These were then transformed into (E)-1g and (Z)-1g (soiled with 10% each of the other stereoisomer) in good yields on reaction with NaH (4 eq., THF, 22 °C, yields: 88% and 89%, respectively). Each mixture (90/10) was further purified by HPLC, as disclosed above.
Synthesis of tritium radiolabeled 2,3-oxidopolyenes
Radiolabeled 2,3-oxidosqualene analogs 1d* (967 dpm/nmol), 1e*(610 dpm/nmol), and 1f* (547 dpm/nmol) were synthesized from the α-tritiated aldehyde 8* using the protocol described above. The latter was generated on heating 8 with T2O in the presence of triethylamine (50 °C, 15 h) and directly used after usual workup.
A different strategy was used for the synthesis of radiolabeled 1c* (2508 dpm/nmol) as model and 1g to avoid the manipulation of radiolabeled material during the tedious separation of the stereoisomers. It was achieved on each stereoisomer after their separation [Scheme 9].
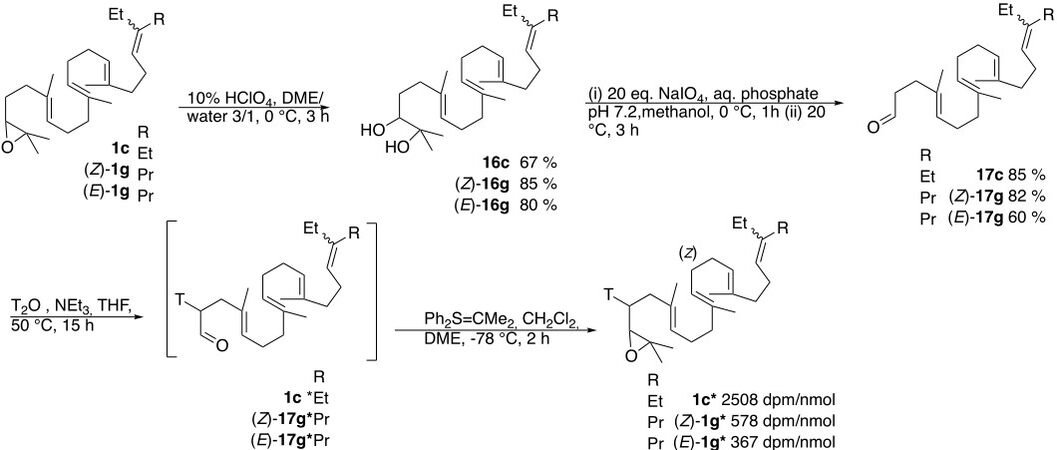
Scheme 9. Transformation of 2,3-oxidosqualenes 1c and 1g into their tritium-labeled analogs at C-4.
Accordingly, 1c and each 1g stereoisomer were degraded to the corresponding aldehyde by acid catalyzed epoxide ring opening leading to the diol 16 (70% HClO4, DME/water 3/1, 0 °C, 3 h) and then transformed to the aldehyde 17 (20 eq. NaIO4, aqueous phosphate pH 7.2, methanol, 0 °C, 1 h, 20 °C, 3 h), tritiated as disclosed above for other aldehydes, and the resulting 17* was reacted with an excess of isopropylidene triphenylsulfurane to deliver radiolabeled epoxide 1c, (Z)-1g, and (E)-1g [(i) 35 eq. isopropyl diphenyl sulfonium tetrafluoroborate, 35 eq. CH2Cl2, DME, -78 °C, 0.5 h; (ii) 1 eq. 17*, DME, -78 °C, 2 h; and (iii) H2O leading to 1c* 48%, 2508 dpm/nmol; (Z)-1g* 44%, 578 dpm/nmol; and (E)-1g* 45% 367 dpm/nmol].
Reaction of 2,3-oxidopolyenes with OSC-PL
The oxidosqualene analogs were reacted with OSC from pig liver prepared according to Hogeboom[19]. The conditions used proved to efficiently cyclize 2,3-oxidosqualene (E)-1a to lanosterol (R)-3a in up 80% yield (40% since the reaction was carried out on epimeric mixture of (E)-1a at C-3).
Accordingly, the radiolabeled 2,3-oxidopolyene 1 (300-2000 nmol) mixed with Tween 80 [10% in acetone (10%) and then evaporation of acetone] and dissolved in bi-distilled water was stirred under argon for 3 h (17 h in the large-scale experiment, 5 mg of 1) with an aqueous solution of OSC-PL prepared as discussed above[19]. The mixture was then saponified on stirring with a 10% potassium hydroxide solution in ethanol and extracted with ether, leading to a recovery of compounds possessing 88%-95% of the radioactivity. The crude mixture was separated by preparative layer chromatography (SiO2, toluene/ethyl acetate 95/05, and the following products were isolated: recovered oxidosqualene 1, rf = 0.7-0.8; lanosterol, rf = 0.4-0.5; or analogs 3, rf = 0.1-0.2). In the case of 1g, the reactions were carried out at 1 μmol, and the purifications on SiO2 (toluene/ethyl acetate 95/05) led to: from (Z)-1g, Zone AZ, rf = 0.7-0.8 (61%, oxidosqualene); Zone BZ, rf = 0.4-0.5 (54%, lanosteryl derivatives); and Zone CZ, rf = 0.1-0.2 (11%, unknown); and, from (E)-1g, Zone AZ, rf = 0.6-0.8 (53%, oxidosqualene), and Zone BZ, rf = 0.4-0.45 (43%, lanosteryl derivatives).
Hemi-synthesis of lanosteryl analogs (R)-3g and (R)-3g
To unambiguously prove the structure of the compounds resulting from the reaction of 1g with 2,3-oxidosqualene sterol cyclase, we achieved the hemi-synthesis of the two epimeric (20R)-3g and (20S)-3g from lanosterol 1a as a common intermediate using a methodology previously used in our laboratory[28,29].
The synthetic strategy involves the following [Schemes 10 and 11]:
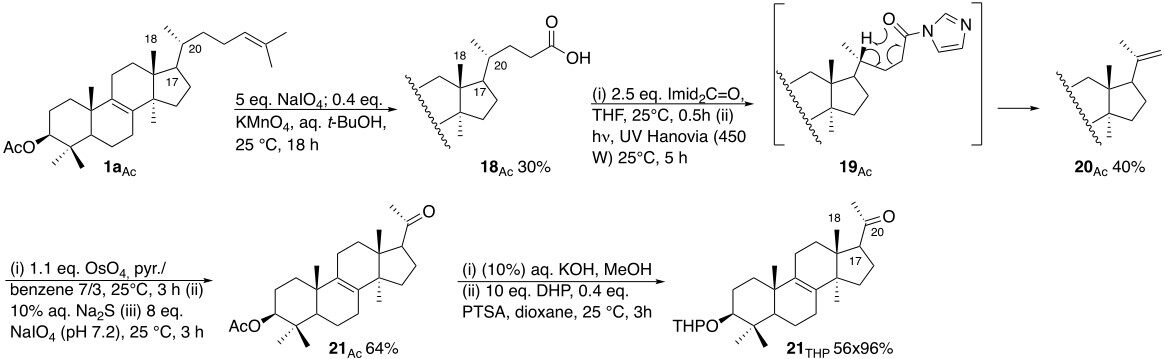
Scheme 10. Description of the synthetic scheme allowing the degradation of the side chain of lanosterol up to C-20.
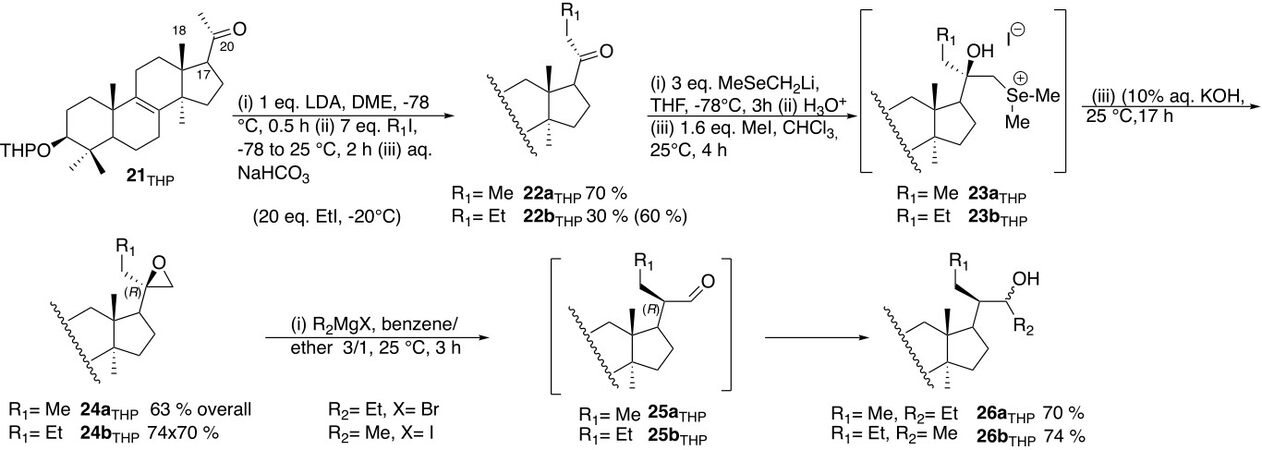
Scheme 11. Stereoselective introduction of asymmetric C-20 carbon precursors of lanosteryl analogs (R)-3g and (S)-3g.
(i) Degrade the lanosteryl acetate 1aAc side chain up to C-20, leading to the methyl ketone 21 in order to destroy the stereochemical information there keeping intact the structure and stereochemistry of the tetracyclic scaffold [Scheme 10][30].
(ii) Restore the chirality at C-20 by taking advantage of [Scheme 11]: (a) the face-selective addition of nucleophile on the (Si)-face of the carbonyl of the ketones 22 directed by the presence of the C-18 methyl group using a nucleophile bearing a leaving group or a potential leaving group able to produce the epoxides 24 [Scheme 9]; and (b) rearrangement of the epoxide 24 to the intermediate aldehyde 25 on reaction with Grignard reagents that involves the highly stereoselective migration of a hydrogen of the epoxide ring leading mainly to the (20R)-epimers of 25 (R = Me or Et) that are trapped in situ by the Grignard reagents.
(iii) Synthesize both epimeric derivatives (20R)-3g and (20S)-3g that involve the electrophilic alkylation (R1-X, X = halogen, R1 = Me, Et)[31] of the methyl ketone 21 and later the nucleophilic alkylation (R2-MgX, R2 = Et, Me) of the aldehyde 25 using the same series of reactions but inverting the sequential introduction of methyl and ethyl groups to access either to (20R)-3g (R1 = Me, R2 = Et) through (22a and 26a, Scheme 11) or to (20S)-3g (R1 = Et, R2 = Me) through (22b and 26b, Scheme 11).
A transformation related to that of 26 to 3g was originally performed in our laboratory[28], and is disclosed in Scheme 12, entry a. It allows the synthesis of (R)-3g through the oxidation of 26bTHP and then acid catalyzed deprotection leading to 27b. The former reaction was readily achieved by using excess of PCC in the presence of sodium acetate (5 eq. PCC, 0.3 eq. AcONa, CH2Cl2, 25 °C, 3 h)[32]. However, we experienced unexpected drawbacks in the synthesis of the thioacetal 28b from 27b. We could not extend to 27b the method used (HS-CH2-CH2-SH, BF3.Et2O, AcOH, 25 °C) on a closely related ketone[30]. Neither alternative methods disclosed by Evans et al.[33] such as (Me3Si-S-CH2-CH2-S-SiMe3, ZnI2, Et2O, 24 h) or the more conventional ones [HS-CH2-CH2-SH (15 eq.), ZnCl2 (8 eq.), CH2Cl2, 25 °C, 8 h] proved to be successful to reach our goal. The latter reaction however, when carried out for longer time (48 h instead of 8 h), delivered the thioacetal 28b in modest yield (51%, Scheme 9). Reduction of the latter with lithium in ethylamine [(i) 250 eq. Li, EtNH2, -25 °C, 0.7 h; and (ii) aq. NH4Cl] affords (R)-3g in 43% yield along with (S)-3g (15%) and one unidentified product.
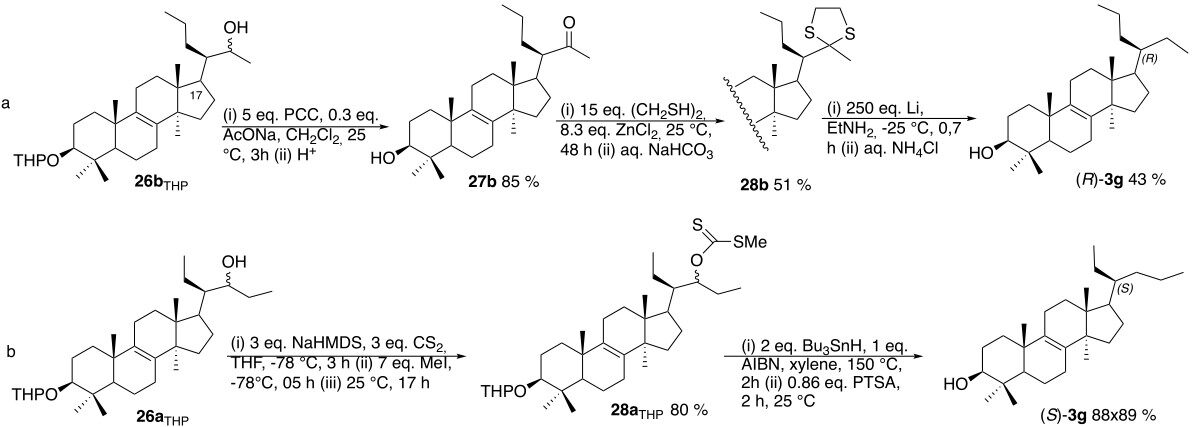
Scheme 12. Final steps towards the synthesis of lanosteryl analogs (R)-3g and (S)-3g.
Due to the difficulty encountered in the transformation disclosed above, we decided to adopt a different strategy to transform 26a into (S)-3g (Scheme 12, entry b) that involves the formal reduction of the alcohol 26aTHP. It was efficiently achieved by modification of the well-known Barton-McCombie method[34] that uses the reduction of the related xanthate.
The synthesis of the xanthate 28aTHP was readily achieved [(i) 3 eq. NaHMDS, 3 eq. CS2, THF, -78 °C, 3 h; (ii) 7 eq. MeI, -78 °C, 05 h; and (iii) 25 °C, 17 h, 80%], but neither its reduction with tin hydride in the presence of azo-bis-isobutyronitrile (AIBN) as a radical initiator (1.5 eq. Bu3SnH, xylene, reflux, 15 h) according to the original procedure[34] nor those involving 2 eq. Bu3SnH and 1 eq. AIBN in toluene (110 °C, 2 h, 10% yield) worked properly. However, we found that performing the reaction at high temperature but for a short time to avoid the degradation of the product proved to be an excellent alternative (2 eq. Bu3SnH, 1 eq. AIBN, xylene, 150 °C, 2 h, 88%).
Both alcohols (20R)-3g and (20S)-3g were acetylated (Ac2O, pyridine, 25 °C, 17 h), delivering (20R)-3gAc and (20S)-3gAc in 84% yield. Those acetates proved to contain 10% of the other diastereoisomer (15% and 10%, respectively), probably arising from the rearrangement of the epoxide with the Grignard reagent step that is less stereoselective than expected[28,29].
CONCLUSIONS
We report the original behavior of OSC-PL towards oxidosqualene analogs bearing two alkyl groups at C19 by the incremental addition of a methyl group from: (a) Me,Et (tetracyclic, stereospecific)[16,17]; (b) Et,Et (tetracyclic); (c) Et,Pr (tetracyclic, non-stereospecific); and (d) Pr;Pr (no cyclization). It adds to the previous work that includes: (e) Me, isohexenyl (tetracyclic/tricylic non-chemospecific)[18]; (f) saturated C18-C19 bond (tricyclic)[20]; and (g) Me, isohexadienyl (tetracyclic, protosterol)[14]. We observed the exceptional ability of this enzyme to respond in a different manner to different substrate analogs, providing each time products possessing a different type of structure.
We also propose an original hypothesis to rationalize the formation for the first time of a lanosterol analog bearing the natural stereochemistry at C-20 from an oxidosqualene analog bearing a non natural stereochemistry at Δ18-19. A possible support for this hypothesis could arise from reacting OSC-PL with an oxidosqualene analog bearing one [Figure 1, (Z)-1h or (Z)-1i] or two unsaturations [Figure 1, (Z)-1j] on the side chain hoping that protosterols bearing the side chain lying in alpha-position would be isolated using a strategy similar to the one developed by Corey et al.[14] using (Z)-1k [Figure 1] to support his proposal reported in Scheme 2, entry b[14,15]. Modeling in silico the process is another alternative that we are pursuing using the detailed X-ray structure of human sterolcyclase reported by Thoma et al.[5].
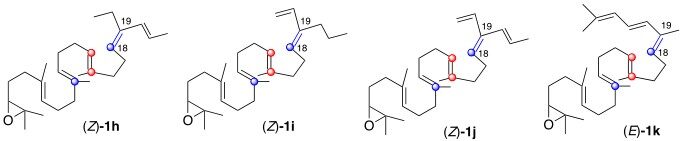
Figure 1. Potential precursors of protesterols bearing a side-chain lying in alpha-position.
DECLARATIONS
AcknowledgmentsThe authors acknowledge the fundamental technical assistance of M. Alain Burlet and of Mrs Anne-Marie Laval. Alain Krief is grateful to Dr. Lucy Stark for the extremely useful translation of this master piece scientific paper - Revisited after 50 Years: The “Stereochemical Interpretation of the Biogenetic Isoprene Rule for the Triterpenes”.
Authors’ contributionsMade similar contributions to conception and design of the study and performed data analysis and interpretation: Krief A, Sable R, Ronvaux A, Dumont W, Sandra P, David F
Availability of data and materialsData are deposited at the Université of Namur in the PhD theses of Dr. Alain Ronvaux (1998) and Dr. Romuald Sable (2007) and are accessible on request.
Financial support and sponsorshipThis work has been supported by doctoral fellowships to Alain Ronvaux and to Romuald Sable from the FRIA (Fund for Research Training in Industry and Agriculture) from the Belgian Science Foundation (FNRS). The authors thank FNRS and the Facultés Universitaires Nôtre Dame de la Paix (FUNDP) for their financial support. Alain Krief deeply thanks Professor Charles Jaumotte previous General Admministrator of the University N. D de la Paix, Namur and the members of its Administration Council for generous financial support and encouragements.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationWritten informed consent for publication was obtained for all the authors and from Dr. Willy Dumont wife (Mrs Josiane Gerlache) and one of his sons (Professor Patrick Dumont, UCL, Louvain le Neuve, Belgium).
Copyright© The Author(s) 2021.
REFERENCES
1. Barrett A, Ma T, Mies T. Recent developments in polyene cyclizations and their applications in natural product synthesis. Synthesis 2018;51:67-82.
2. Yoder RA, Johnston JN. A case study in biomimetic total synthesis: polyolefin carbocyclizations to terpenes and steroids. Chem Rev 2005;105:4730-56.
3. Huff MW, Telford DE. Lord of the rings - the mechanism for oxidosqualene:lanosterol cyclase becomes crystal clear. Trends Pharmacol Sci 2005;26:335-40.
5. Thoma R, Schulz-Gasch T, D'Arcy B, et al. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature 2004;432:118-22.
6. Woodward RB, Bloch K. The cyclization of squalene in cholesterol synthesis. J Am Chem Soc 1953;75:2023-4.
7. Stork G. The stereochemistry of polyene cyclization. Harvard University Organic colloquium Abstract. 1950. Burgstahler AW, PhD. Thesis. Harvard University: 1952.
8. Stork G, Burgstahler AW. The stereochemistry of polyene cyclization. J Am Chem Soc 1955;77:5068-77.
9. Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Zur Kenntnis der Triterpene. 190. Mitteilung. Eine stereochemische Interpretation der biogenetischen Isoprenregel bei den Triterpenen. Helv Chim Acta 1955;38:1890-904.
10. Eschenmoser A, Arigoni D. Revisited after 50 years: the “stereochemical interpretation of the biogenetic isoprene rule for the triterpenes”. HCA 2005;88:3011-50.
11. Corey EJ, Russey WE, Ortiz de Montellano PR. 2,3-Oxidosqualene, an intermediate in the biological synthesis of sterols from squalene. J Am Chem Soc 1966;88:4750-1.
12. Tamelen EE, Willett JD, Clayton RB, Lord KE. Enzymic conversion of squalene 2,3-oxide to lanosterol and cholesterol. J Am Chem Soc 1966;88:4752-4.
14. Corey EJ, Virgil SC, Sarshar S. New mechanistic and stereochemical insights on the biosynthesis of sterols from 2,3-oxidosqualene. J Am Chem Soc 1991;113:8171-2.
15. Wendt KU, Schulz GE, Corey EJ, Liu DR. Enzyme mechanisms for polycyclic triterpene formation. Angew. Chem. Int. Edn Engl 2000; 39:2812-33.
16. Hérin M, Sandra P, Krief A. Stereospecific enzymic cyclization of a synthetic 2,3-oxidosqualene analogue bearing an 18Z carbon-carbon double bond. Tetrahedron Letters 1979;20:3103-6.
17. Krief A, Pasau P, Guittet E, Shan YY, Hérin M. Novel results on the biocyclisation of 2,3-oxidosqualene analogs by sterol cyclase. Bioorganic & Medicinal Chemistry Letters 1993;3:365-8.
18. Krief A, Schauder JR, Guittet E, Herve du Penhoat C, Lallemand JY. About the mechanism of sterol biosynthesis. J Am Chem Soc 1987;109:7910-1.
19. Hogeboom GH. Methods enzymol. Fractionation of cell components of animal tissues. Elsevier; 1955. p. 16-9.
20. Tamelen EE, Sharpless KB, Hanzlik R, Clayton RB, Burlingame AL, Wszolek PC. Enzymic cyclization of trans,trans,trans-18,19-dihydrosqualene 2,3-oxide. J Am Chem Soc 1967;89:7150-1.
21. Krief A, Pasau P, Quéré L. Comparison of the behavior of oxidosqualene cyclases from pig liver and yeast toward epoxy-squalene analogues possessing a Δ18-19 Z or E (C, C) double bond. Bioorganic & Medicinal Chemistry Letters 1991;1:365-8.
22. Krief A, Dunkle M, Bahar M, Bultinck P, Herrebout W, Sandra P. Elucidation of the absolute configuration of rhizopine by chiral supercritical fluid chromatography and vibrational circular dichroism. J Sep Sci 2015;38:2545-50.
23. Tamelen E, Sharpless KB. Positional selectivity during controlled oxidation of polyolefins. Tetrahedron Letters 1967;8:2655-9.
24. Krief A, Dumont W, Clarembeau M, Bernard G, Badaoui E. Synthesis of α-selenoalkyllithium compounds. Tetrahedron 1989;45:2005-22.
25. Labar D, Krief A. Connective (C-C) route to hindered epoxides and olefins from hindered ketones. J Chem Soc, Chem Commun 1982;10:564-6.
26. Horner L, Hoffmann H, Wippel HG, Klahre G. Phosphororganische Verbindungen, XX. Phosphinoxyde als Olefinierungsreagenzien. Chem Ber 1959;92:2499-505.
27. Buss AD, Warren S. The Wittig-Horner route to tri-substituted alkanes: synthesis of -α-bisabolene. Tetrahedron Letters 1983;24:111-4.
28. Schauder J, Krief A. Regio and stereochemically controlled ring opening of epoxides with Grignard reagents. Stereocontrolled synthesis of the steroid side chains. first stereoselective hemisynthesis of 20s isolanosterol. Tetrahedron Letters 1982;23:4389-92.
29. Koreeda M, Koizumi N. Stereochemically controlled synthesis of 20-isocholesterol. Tetrahedron Letters 1978;19:1641-4.
30. Pasau P. PhD thesis, Facultés Universitaires Notre-Dame de la Paix (FUNDP). Namur; 1992.
31. House HO, Gall M, Olmstead HD. Chemistry of carbanions. XIX. Alkylation of enolates from unsymmetrical ketones. J Org Chem 1971;36:2361-71.
32. Corey EJ, Suggs JW. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Letters 1975;16:2647-50.
33. Evans DA, Truesdale LK, Grimm KG, Nesbitt SL. Thiosilanes, a promising class of reagents for selective carbonyl protection. J Am Chem Soc 1977;99:5009-17.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Krief A, Sable R, Ronvaux A, Dumont W, Sandra P, David F. Schizophrenic behavior of 2,3-oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs. Chem Synth 2021;1:6. http://dx.doi.org/10.20517/cs.2021.03
AMA Style
Krief A, Sable R, Ronvaux A, Dumont W, Sandra P, David F. Schizophrenic behavior of 2,3-oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs. Chemical Synthesis. 2021; 1(1): 6. http://dx.doi.org/10.20517/cs.2021.03
Chicago/Turabian Style
Krief, Alain, Romuald Sable, Alain Ronvaux, Willy Dumont, Pat Sandra, Frank David. 2021. "Schizophrenic behavior of 2,3-oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs" Chemical Synthesis. 1, no.1: 6. http://dx.doi.org/10.20517/cs.2021.03
ACS Style
Krief, A.; Sable R.; Ronvaux A.; Dumont W.; Sandra P.; David F. Schizophrenic behavior of 2,3-oxidosqualene sterol cyclase from pig liver towards 2,3-oxidosqualene analogs. Chem. Synth. 2021, 1, 6. http://dx.doi.org/10.20517/cs.2021.03
About This Article
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 35
likes
Like This Article 35
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.