
Enantioselective 1,1-diarylation of terminal alkenes catalyzed by palladium with a chiral phosphoric acid


Abstract
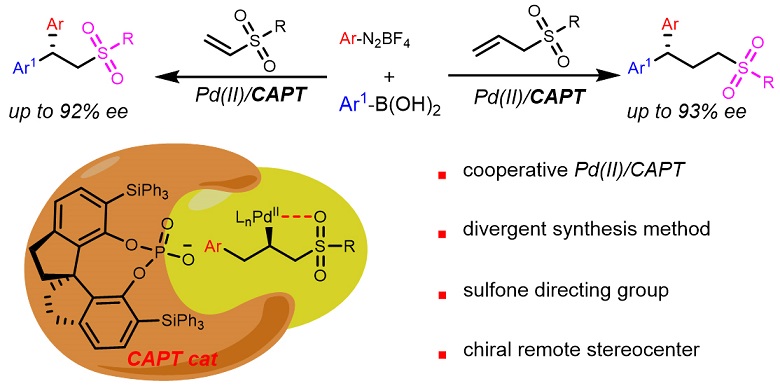
The enantioselective 1,1-diarylation of allyl sulfones and vinyl sulfones is reported for the first time, enabled by a combination of Pd2dba3 and a chiral SPINOL-derived phosphoric acid. Various chiral sulfones containing 1,1-diarylalkane motifs were obtained in moderate to good yields with moderate to high enantioselectivities. Control experiments suggested that the sulfone group plays a key role in providing enantioselectivity and reactivity control and might serve as a directing group.
Keywords
INTRODUCTION
The chiral 1,1-diarylalkane motif is found in many bioactive natural products and pharmaceuticals
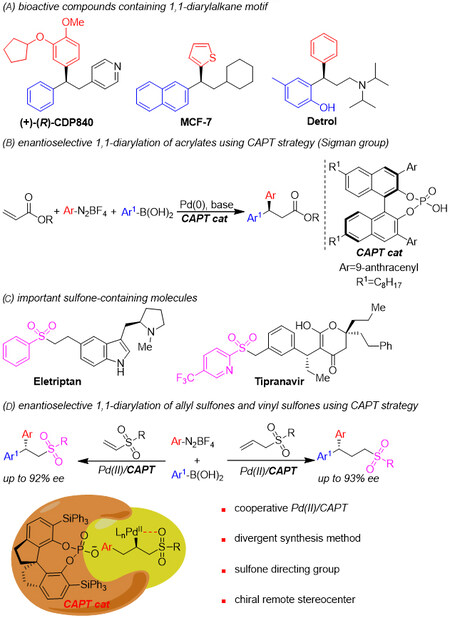
Scheme 1. Design of the enantioselective 1,1-diarylation of allyl sulfones and vinyl sulfones.
Sulfone groups are widely found in biologically active molecules [Scheme 1C][18-21]. Compounds containing these groups are also versatile synthetic intermediates because sulfones are easily transformed into a range of structurally diverse functional groups and can also function as flexible directing groups in various metal-catalyzed reactions to provide site- and enantioselective processes and control reactivity[22-26]. On this basis, we considered that sulfone groups might permit a high level of selectivity during catalytic asymmetric 1,1-diarylation. We aimed to synthesize the highly enantioselective 1,1-diarylation of allyl sulfones. This process involved two main challenges. First, allyl sulfones are less reactive than activated alkenes and so competitive reactions become more important. Examples include the traditional Heck reactions of alkene substrates and aryldiazonium salts[27-30] and the Suzuki reaction of aryldiazonium salts and arylboronic acids[16,31]. Second, the enantioselectivity control of this novel reaction process was expected to be more difficult. For these reasons, it was thought that a new catalysis system might be required. Herein, we report the highly enantioselective 1,1-diarylation of allylic sulfones using a new co-catalysis system comprising a chiral SPINOL-derived phosphoric acid and Pd2dba3. This methodology allows modular and direct access to chiral sulfones that contain a 1,1-diarylalkane motif and a remote stereocenter [Scheme 1D]. This new co-catalysis system was also found to be applicable to vinyl sulfones.
EXPERIMENTAL
In glovebox, 2a (0.2 mmol), 3a (0.2 mmol), 4a (0.1 mmol), Pd2dba3 (3.7 mg, 0.004 mmol), (R)-1a (10.0 mg, 0.012 mmol), Rb2CO3 (69.3 mg, 0.3 mmol), anhydrous MTBE (2.0 mL) were added into an oven-dried tube (10 mL) charged with a stir bar, sealed the flask with its septum and then removed from the glovebox. The system was stirred for 48 h at 0 ºC. After the reaction was completed (monitored by TLC), the crude reaction mixture was filtered through a short silica column with PE: EA = 1:2, and the solvent was removed in vacuo. An external standard (dibromomethane) was added to the system for determining NMR yields. Product 5a was obtained by PTLC.
RESULTS AND DISCUSSION
In our initial study, allyl sulfone 2a was chosen as a model substrate, while 4-methoxybenzene diazonium salt 3a and phenylboronic acid 4a were selected as model coupling partners. A series of careful screening trials were performed (see Supplementary Materials for details). As indicated in Scheme 2 and Table 1, entry 1, the desired product 5a was obtained in 64% yield with 96:4 er when Pd2dba3 and chiral SPINOL-derived phosphoric acid (R)-1a [Figure 1] were used as the catalysts in the presence of Rb2CO3 in methyl tert-butyl ether (MTBE) at 0 ºC. The use of a chiral phosphoric acid was vital to obtaining the desired reactivity and enantioselectivity during this transformation. When (R)-1b bearing a phenyl substituent was used instead of (R)-1a, 5a was obtained in only 11% yield with 71:29 er (entry 2), with the remainder being the traditional Heck product. Replacing (R)-1a with chiral BINOL-derived phosphoric acid (R)-1c resulted in a reduction in both the yield and enantioselectivity (entry 3). It was found that the absolute configuration of product 5a was reversed, which met the opposite axial chirality of (R)-BINOL 1c and (R)-SPINOL 1a. Similar results were obtained using (R)-1e (entry 5). Both lower yield and reduced enantioselectivity were observed when chiral BINOL-derived phosphoric acid (R)-1d and H8-BINOL-derived (R)-1f were used (entries 4, 6) and the primary result was again the traditional Heck product. A series of different inorganic bases was subsequently examined in place of Rb2CO3 (entries 7-9). These trials indicated that the particular base that was used had a significant effect on the yield but only slightly modified the enantioselectivity. Using K2CO3 as a base gave similar results as obtained with the standard conditions, while employing Cs2CO3 or Na2CO3 led to an obvious reduction in yield. Using diethyl ether as the solvent, the enantioselectivity was lowered but not the yield (entry 10). Only a trace amount of 5a was obtained when toluene was substituted as the solvent (entry 11). Product 5a was obtained in 21% yield with 88:12 er when this reaction was performed at room temperature (entry 12).
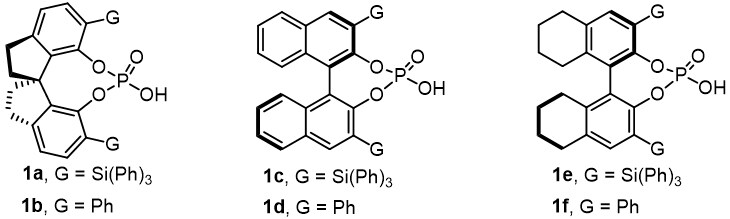
Figure 1. Some different chiral phosphoric acids.
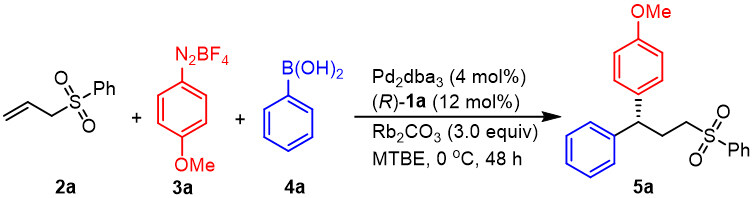
Scheme 2. The standard conditions of the catalytic asymmetric 1,1-diarylation of allylic sulfone.
Reaction optimizationa
| Entry | Change from the “standard conditions” | Yield (%)b | erc |
| 1 | none | 64 | 96:4 |
| 2 | (R)-1b instead of (R)-1a | 11 | 71:29 |
| 3 | (R)-1c instead of (R)-1a | 10 | 13:87 |
| 4 | (R)-1d instead of (R)-1a | 12 | 65:35 |
| 5 | (R)-1e instead of (R)-1a | 24 | 12:88 |
| 6 | (R)-1f instead of (R)-1a | 27 | 71:29 |
| 7 | Cs2CO3 instead of Rb2CO3 | 51 | 96:4 |
| 8 | K2CO3 instead of Rb2CO3 | 62 | 95:5 |
| 9 | Na2CO3 instead of Rb2CO3 | 44 | 94:6 |
| 10 | Et2O instead of MTBE | 66 | 90:10 |
| 11 | toluene instead of MTBE | trace | - |
| 12 | rt instead of 0 ºC | 21 | 88:12 |
Taking advantage of the optimized conditions, the reaction scope was first examined with a variety of arylboronic acids. In general, all arylboronic acids gave the desired product in a moderate yield with moderate to high levels of enantioselectivity [Scheme 3]. We found that the presence of methyl or fluorine groups at the 4-position of the phenyl ring slightly affected the yield and enantioselectivity, while electron-deficient substituents such as chlorine, ester and trifluoromethyl groups resulted in decreased yields and enantioselectivities (5b-5f). The reaction was also found to proceed with (4-hydroxyphenyl) boronic acid, giving the desired product 5g in 96.5:3.5 er, albeit with a yield of 27%. Following these trials, phenylboronic acids bearing meta substituents such as methoxy and methyl groups were also applied to this reaction, and the corresponding products were obtained with moderate yields and high enantioselectivities (5h-5i). Interestingly, ortho-tolylboronic acid also worked well in this protocol, yielding product 5j in 62% yield and 96:4 er. A multi-substituted phenylboronic acid was applied to this system and provided product 5k with a high level of enantioselectivity. Naphthalen-2-ylboronic acid was also a suitable coupling partner and delivered product 5l in 53% yield with 95.5:4.5 er. It is worth noting that a series of (heter)arylboronic acids were also examined in this reaction. In these trials, product 5m was obtained in 60% yield with 86.5:13.5 er, while products 5n and 5o were generated in lower yields with moderate enantioselectivities (More results about (hetero)arylboronic acids, please see SI). Following the experiments described above, the reaction scope was examined by assessing different aryldiazonium salts (5p-5s). Changing the substituent of the model coupling partner 3a from a methoxy to a benzyloxy group maintained high enantioselectivity and also gave a moderate yield. It should be noted that the absolute configuration of 5p was assigned by X-ray crystallography[32]. It was also determined that the meta-methoxybenzene diazonium salt was compatible with this reaction, giving product 5q with 40% yield and 95.5:4.5 er. In addition, using ethyl ether as the solvent, product 5q was produced in 53% yield with a lower er value. In further trials, para-tolylboronic acid and naphthalen-2-ylboronic acid were employed as coupling partners, and the corresponding products 5r and 5s were generated in moderate yields with good enantioselectivities. As expected, when the phenyl diazonium salt and 4-methylboronic acid were used as coupling partners, product ent-5r was obtained in 40% yield with 11:89 er. These results indicate that the range of aryldiazonium salts applicable to this process is relatively limited.
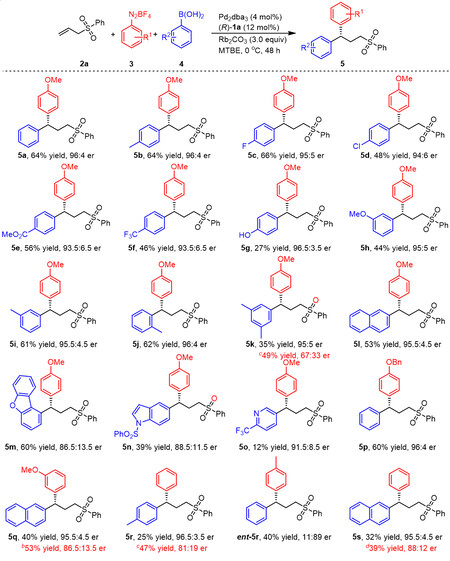
Scheme 3. Scope of arylboronic acids and aryldiazonium saltsa. aUnless otherwise noted, the reaction was conducted with 2a
To further extend the scope of this reaction, we subsequently explored various allyl sulfone substrates [Scheme 4]. The steric effects of phenyl ring substituents were initially examined. A methyl group at the 4-position of the phenyl moiety was found not to affect the yield or enantioselectivity, while a tertiary butyl group decreased the yield (5t-5u). The electronic effect of substituents was also evaluated. Both an electron-rich methoxy substituent and electron-poor substituents such as fluorine, ester and trifluoromethyl groups slightly decreased the yield and enantioselectivity (5v-5y). Substrates bearing meta-methoxy-phenyl, 3,5-dichlorophenyl and 1-naphthyl moieties were also examined. In each case, the corresponding product was obtained with moderate efficiency and a high level of enantioselectivity (5z-5ab). However, a substrate with a SO2Py group did not give the desired product (5ac), presumably because the pyridine group strongly coordinated with the palladium to suppress the reaction. Additionally, the use of alkyl groups, such as benzyl and tertiary butyl groups, instead of the phenyl group resulted in significant decreases in yield and enantioselectivity (5ad-5ae). From these data, it is evident that the sulfone group was a vital aspect of obtaining enantioselectivity and reactivity control. To our delight, allyl sulfonates were also suitable for this reaction. The products 5af-5ah were always obtained in moderate yields with high enantioselectivities. As expected, allyl sulfonamide suppressed the transformation.
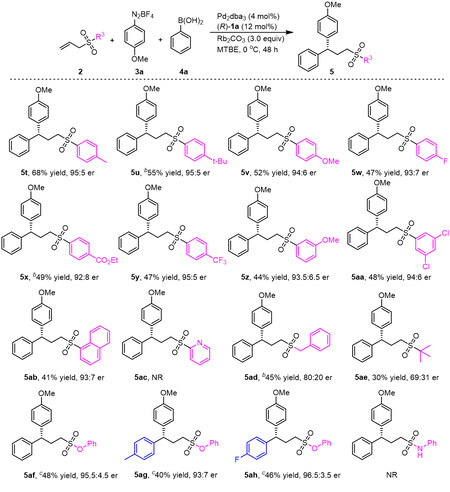
Scheme 4. Scope of allylic sulfonesa. aUnless otherwise noted, the reaction was conducted with 2 (0.2 mmol), 3a (0.2 mmol), 4a
Because chiral β,β-diaryl sulfones can be readily transformed into bioactive pharmaceutical molecules such as (R)-CDP-840 and (R)-MCF-7[33,34], the extension of this catalytic system to the 1,1-diarylation of vinyl sulfones was investigated. The desired product 7a was obtained in a 67% yield and with 95:5 er from the reaction of vinyl sulfone 6a, 4-methoxybenzene diazonium salt 3a and phenylboronic acid 4a, using Cs2CO3 instead of Rb2CO3 [Scheme 5]. Following this, a number of arylboronic acids were applied to the reaction and the corresponding products were obtained in moderate yields with high enantioselectivities (7b-7f). It is of note that ortho-tolylboronic acid was also suitable for this protocol and gave product 7e in 45% yield and 96:4 er. Phenyl aryldiazonium salt was employed in this reaction and product 7g was obtained in 45% yield and 93.5:6.5 er. The absolute configuration of (S)-7g was determined by comparison with optical rotation data for the reported literature[26]. Likewise, the product 7h was obtained in 30% yield with 96:4 er using vinyl sulfonate as the substrate. Finally, product ent-7a was produced in moderate yield with good enantioselectivity.
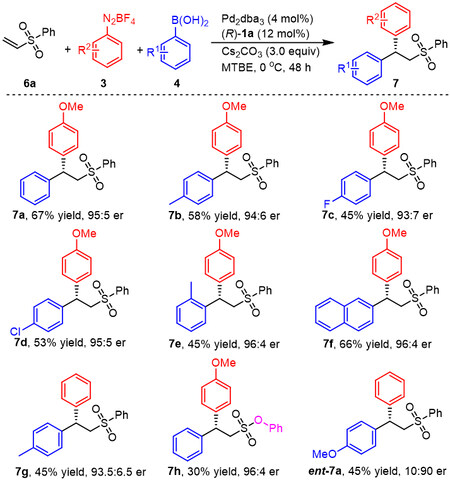
Scheme 5. Scopes of arylboronic acids and aryldiazonium saltsa. aUnless otherwise noted, the reaction was conducted with 6a
The origin of the high enantioselectivities obtained from this reaction and the effect of the sulfone group were explored by carrying out the reaction using several different substrates [Scheme 6A]. These trials showed that increasing the length of the carbon chain between the sulfone group and the double bond greatly affected the yield and enantioselectivity (5ai-5aj). The reaction was also inhibited upon changing from a sulfone group to either a sulfoxide or thioether group (5ak, 5al). Additionally, the reaction was suppressed after substituting a sulfoxide or thioether group for the sulfone (5ak, 5al), possibly because of the strong coordination effects of the sulfoxide and thioether groups. When but-3-en-1-ylbenzene was used instead of 2a, product 5am was only obtained in 17% yield and 72:28 er. On the basis of these results, we suggest that the sulfone moiety serves as a directing group during this reaction. An internal alkene substrate was also tested, and the desired product 5an, bearing a quaternary carbon center, was obtained in only a 20% yield with just a 63.5:36.5 er value. Thus, a terminal double bond is essential for high enantioselectivity. The effects of the boronic acid group on the reactivity and enantioselectivity of this process were first assessed by using esterified phenylboronic acid 9a instead of phenylboronic acid and no desired product 5a was obtained [Scheme 6B]. Product 5a was obtained in 37% yield with 88:12 er [Scheme 6C]. The negative results established that the standard boronic acid group was crucial to this reaction. The use of Pd(OAc)2 instead of Pd2dba3 as the catalyst afforded the desired product 5a with 96:4 er, suggesting that the dba moieties did not participate in the asymmetric catalytic reaction [Scheme 6D]. Because the mass balance was the traditional Heck product 8a in this transformation, this compound was employed as the starting material in a subsequent trial with the standard reaction conditions. The desired product 5a was not obtained [Scheme 6E]. As shown in Scheme 6F, a cross-over experiment was further performed, but no cross-over product was obtained. Based on these observations, we believe that the Heck product was unable to participate in subsequent reactions with the arylboronic acid to form the 1,1-diarylation product.
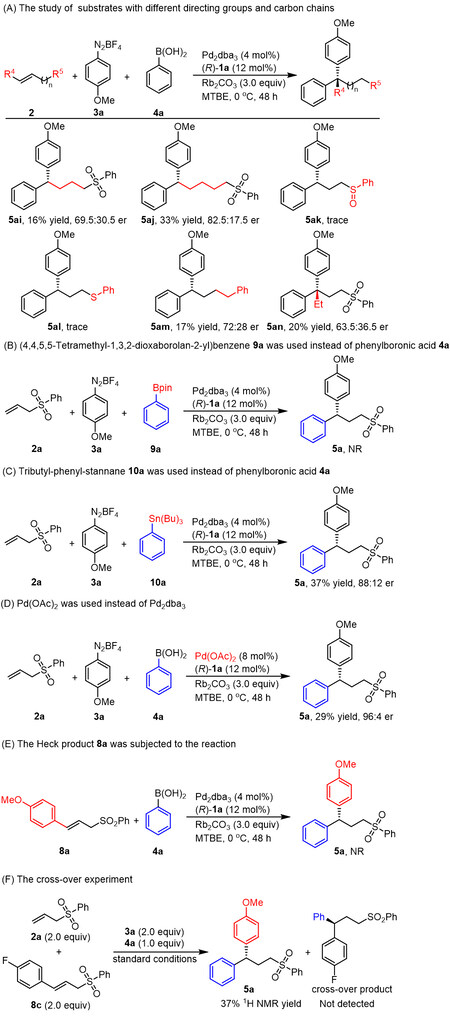
Scheme 6. Mechanistic studies.
Based on these experimental results and previous work[10], a possible mechanism for the catalytic cycle was devised [Scheme 7]. In this mechanism, soluble chiral ion pair A is first generated from a chiral phosphate anion and the insoluble tetrafluoroborate aryldiazonium salt in the presence of Rb2CO3. Cationic Pd-aryl intermediate B is then formed via the oxidative addition of A in conjunction with Pd2dba3 and subsequently undergoes an enantioselective migratory insertion of 2a to form intermediate C. Intermediate E is generated from C through β-hydride elimination and reinsertion. Finally, E reacts with phenylboronic acid 4a, followed by reductive elimination, forming 1,1-diarylation product 5a.
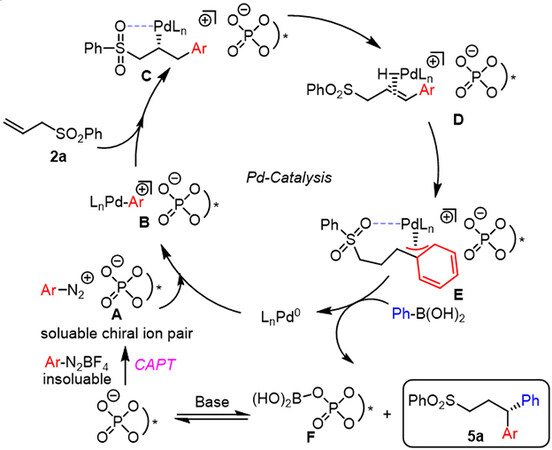
Scheme 7. Proposed catalytic cycle. CAPT: Chiral anion phase-transfer catalysis.
CONCLUSION
In conclusion, this work demonstrated the enantioselective 1,1-diarylation of allyl sulfones using a cooperative strategy based on a chiral SPINOL-derived phosphoric acid and Pd2dba3. This newly developed cooperative catalysis was also applied to the asymmetric 1,1-diarylation of vinyl sulfones. This catalytic system allows for the rapid construction of a variety of chiral sulfones containing a 1,1-diarylalkane motif and a remote stereocenter. Interestingly, we discovered that the sulfone group can function as a directing group and is important for achieving enantioselectivity and reactivity control. The investigation of detailed mechanisms and cooperative catalysis as a means of synthesizing other useful compounds is ongoing in our laboratory.
DECLARATIONS
AcknowledgmentsWe gratefully thank Prof. Changkun Li and Prof. Xiaohong Huo, Yicong Luo (Shanghai Jiao Tong University) for helpful suggestions and comments on this manuscript.
Authors’ contributionsDesigning the experiments, writing the manuscript, and being responsible for the whole work: Chen ZM
Performing the experiments: Ji K, Huang J
Synthesizing the substrates: Ji K, Huang J, Zhang XY
Availability of data and materialsNot applicable.
Financial support and sponsorshipWe thank the NSFC (grant nos. 21871178 and 22071149) and the STCSM (grant no. 19JC1430100) for financial support. This research was also supported by The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Ameen D, Snape TJ. Chiral 1,1-diaryl compounds as important pharmacophores. Med Chem Commun 2013;4:893.
2. Malhotra B, Gandelman K, Sachse R, Wood N, Michel MC. The design and development of fesoterodine as a prodrug of 5-hydroxymethyl tolterodine (5-HMT), the active metabolite of tolterodine. Curr Med Chem 2009;16:4481-9.
3. Wang H, Tong HR, He G, Chen G. An enantioselective bidentate auxiliary directed palladium-catalyzed benzylic C-H arylation of amines using a BINOL phosphate ligand. Angew Chem Int Ed Engl 2016;55:15387-91.
4. Zhang W, Wu L, Chen P, Liu G. Enantioselective arylation of benzylic C-H bonds by copper-catalyzed radical relay. Angew Chem Int Ed Engl 2019;58:6425-9.
5. Wu L, Wang F, Wan X, Wang D, Chen P, Liu G. Asymmetric Cu-catalyzed intermolecular trifluoromethylarylation of styrenes: enantioselective arylation of benzylic radicals. J Am Chem Soc 2017;139:2904-7.
6. Wang D, Wu L, Wang F, et al. Asymmetric copper-catalyzed intermolecular aminoarylation of styrenes: efficient access to optical 2,2-diarylethylamines. J Am Chem Soc 2017;139:6811-4.
7. Yin Y, Dai Y, Jia H, et al. Conjugate addition-enantioselective protonation of N-aryl glycines to α-branched 2-vinylazaarenes via cooperative photoredox and asymmetric catalysis. J Am Chem Soc 2018;140:6083-7.
8. Belal M, Li Z, Lu X, Yin G. Recent advances in the synthesis of 1,1-diarylalkanes by transition-metal catalysis. Sci China Chem 2021;64:513-33.
9. Li Y, Wu D, Cheng HG, Yin G. Difunctionalization of alkenes involving metal migration. Angew Chem Int Ed Engl 2020;59:7990-8003.
10. Yamamoto E, Hilton MJ, Orlandi M, Saini V, Toste FD, Sigman MS. Development and analysis of a Pd(0)-catalyzed enantioselective 1,1-diarylation of acrylates enabled by chiral anion phase transfer. J Am Chem Soc 2016;138:15877-80.
11. Orlandi M, Hilton MJ, Yamamoto E, Toste FD, Sigman MS. Mechanistic investigations of the Pd(0)-catalyzed enantioselective 1,1-diarylation of benzyl acrylates. J Am Chem Soc 2017;139:12688-95.
12. Mukherjee S, List B. Chiral counteranions in asymmetric transition-metal catalysis: highly enantioselective Pd/Brønsted acid-catalyzed direct alpha-allylation of aldehydes. J Am Chem Soc 2007;129:11336-7.
13. Mahlau M, List B. Asymmetric counteranion-directed catalysis: concept, definition, and applications. Angew Chem Int Ed Engl 2013;52:518-33.
14. Ohmatsu K, Ito M, Kunieda T, Ooi T. Ion-paired chiral ligands for asymmetric palladium catalysis. Nat Chem 2012;4:473-7.
15. Urkalan KB, Sigman MS. Palladium-catalyzed oxidative intermolecular difunctionalization of terminal alkenes with organostannanes and molecular oxygen. Angew Chem Int Ed Engl 2009;48:3146-9.
16. Saini V, Liao L, Wang Q, Jana R, Sigman MS. Pd(0)-catalyzed 1,1-diarylation of ethylene and allylic carbonates. Org Lett 2013;15:5008-11.
17. Li Z, Wu D, Ding C, Yin G. Modular synthesis of diarylalkanes by nickel-catalyzed 1,1-diarylation of unactivated terminal alkenes. CCS Chem 2021;3:576-82.
18. Ilardi EA, Vitaku E, Njardarson JT. Data-mining for sulfur and fluorine: an evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J Med Chem 2014;57:2832-42.
19. Feng M, Tang B, Liang SH, Jiang X. Sulfur containing scaffolds in drugs: synthesis and application in medicinal chemistry. Curr Top Med Chem 2016;16:1200-16.
20. Scott KA, Njardarson JT. Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem (Cham) 2018;376:5.
21. Giannetti AM, Wong H, Dijkgraaf GJ, et al. Identification, characterization, and implications of species-dependent plasma protein binding for the oral Hedgehog pathway inhibitor vismodegib (GDC-0449). J Med Chem 2011;54:2592-601.
22. Tsui GC, Lautens M. Linear-selective rhodium(I)-catalyzed addition of arylboronic acids to allyl sulfones. Angew Chem Int Ed Engl 2010;49:8938-41.
23. Nishimura T, Takiguchi Y, Hayashi T. Effect of chiral diene ligands in rhodium-catalyzed asymmetric addition of arylboronic acids to α,β-unsaturated sulfonyl compounds. J Am Chem Soc 2012;134:9086-9.
24. So CM, Kume S, Hayashi T. Rhodium-catalyzed asymmetric hydroarylation of 3-pyrrolines giving 3-arylpyrrolidines: protonation as a key step. J Am Chem Soc 2013;135:10990-3.
25. Lim KM, Hayashi T. Rhodium-catalyzed asymmetric arylation of allyl sulfones under the conditions of isomerization into alkenyl sulfones. J Am Chem Soc 2015;137:3201-4.
26. Yan Q, Xiao G, Wang Y, Zi G, Zhang Z, Hou G. Highly efficient enantioselective synthesis of chiral sulfones by Rh-catalyzed asymmetric hydrogenation. J Am Chem Soc 2019;141:1749-56.
27. Oxtoby LJ, Gurak JA Jr, Wisniewski SR, Eastgate MD, Engle KM. Palladium-catalyzed reductive heck coupling of alkenes. Trends Chem 2019;1:572-87.
28. Tao ZL, Adili A, Shen HC, Han ZY, Gong LZ. Catalytic enantioselective assembly of homoallylic alcohols from dienes, aryldiazonium salts, and aldehydes. Angew Chem Int Ed Engl 2016;55:4322-6.
29. Zhang T, Li WA, Shen HC, Chen SS, Han ZY. Chiral-anion-mediated asymmetric heck-matsuda reaction of acyclic alkenyl alcohols. Org Lett 2021;23:1473-7.
30. Werner EW, Sigman MS. Operationally simple and highly (E)-styrenyl-selective Heck reactions of electronically nonbiased olefins. J Am Chem Soc 2011;133:9692-5.
31. Zong Y, Hu J, Sun P, Jiang X. Synthesis of biaryl derivatives via a magnetic Pd-NPs-catalyzed one-pot diazotization - cross-coupling reaction. Synlett 2012;23:2393-6.
32. CCDC 2152365 (5p) contains the supplementary crystallographic data for this paper. More detail, see supporting information chapter 6. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre. Available from: www.ccdc.cam.ac.uk/structures [Last accessed on 12 Oct 2022].
33. Lin Y, Kuo Y, Kuei C, Chang M. Palladium-mediated synthesis of 1,1,2-triarylethanes. Application to the synthesis of CDP-840. Tetrahedron 2017;73:1275-82.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ji K, Huang J, Zhang XY, Chen ZM. Enantioselective 1,1-diarylation of terminal alkenes catalyzed by palladium with a chiral phosphoric acid. Chem Synth 2022;2:17. http://dx.doi.org/10.20517/cs.2022.27
AMA Style
Ji K, Huang J, Zhang XY, Chen ZM. Enantioselective 1,1-diarylation of terminal alkenes catalyzed by palladium with a chiral phosphoric acid. Chemical Synthesis. 2022; 2(4): 17. http://dx.doi.org/10.20517/cs.2022.27
Chicago/Turabian Style
Ji, Kai, Jie Huang, Xin-Yu Zhang, Zhi-Min Chen. 2022. "Enantioselective 1,1-diarylation of terminal alkenes catalyzed by palladium with a chiral phosphoric acid" Chemical Synthesis. 2, no.4: 17. http://dx.doi.org/10.20517/cs.2022.27
ACS Style
Ji, K.; Huang J.; Zhang X.Y.; Chen Z.M. Enantioselective 1,1-diarylation of terminal alkenes catalyzed by palladium with a chiral phosphoric acid. Chem. Synth. 2022, 2, 17. http://dx.doi.org/10.20517/cs.2022.27
About This Article
Special Issue
Copyright

Author Biographies




Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 4
likes
Like This Article 4
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.