
A breakthrough in 2-indolylmethanol-involved organocatalytic asymmetric reactions


Keywords
The organocatalytic asymmetric construction of chiral indole-based scaffolds has become an important research field because the wide distribution of such scaffolds in natural products, pharmaceuticals, chiral organocatalysts, and ligands[1]. Among different approaches, 2-indolylmethanols have been recognized as a class of versatile platform molecules in organocatalytic asymmetric transformations for constructing chiral indole-based scaffolds[1-3]. As summarized in Figure 1A, under the catalysis of chiral Brønsted acid (B*-H)[4-5], 2-indolylmethanols readily undergo dehydration to generate carbocation intermediates A-B and vinyliminiums C, which can be illustrated as delocalized cations D. Due to the steric effect of the two R groups (particularly when R is an aryl group), nucleophiles (Nu) more readily attack carbocation B than carbocation A, thus resulting in the C3-umpolung reactivity of 2-indolylmethanols[1-2]. Namely, the C3-position of the indole ring is changed from nucleophilic to electrophilic. Nevertheless, in some cases,
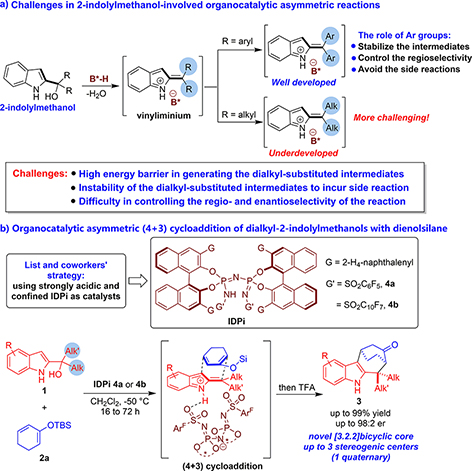
Despite the rapid progress in this research field, there are still some challenges in 2-indolylmethanol-involved organocatalytic asymmetric reactions. As shown in Figure 1B, most of the above-mentioned transformations utilize diaryl-substituted 2-indolylmethanols as reactants. However, in sharp contrast, dialkyl-substituted 2-indolylmethanols have scarcely been utilized as successful reactants in organocatalytic asymmetric reactions. This is because the two aryl groups play important roles in stabilizing the intermediates (vinyliminium and carbocation), controlling the regioselectivity, and avoiding the side reactions. On the contrary, there are great challenges in realizing highly regio- and enantioselective transformations of dialkyl-substituted 2-indolylmethanols, which mainly include: (1) high energy barrier in generating the dialkyl-substituted intermediates; (2) instability of the dialkyl-substituted intermediates to incur side reaction; and (3) difficulty in controlling the regio- and enantioselectivity of the reaction. Therefore, developing innovative strategies to solve these challenges is highly valuable.
To solve these challenging issues, on the basis of their previous studies on silylium-based asymmetric counteranion-directed catalysis (Si-ACDC)[8-9], List and coworkers recently designed an asymmetric (4 + 3) cycloaddition of dialkyl-substituted 2-indolylmethanols 1 with dienolsilane 2a by using IDPi (imidodiphosphorimidates) as strongly acidic and confined organocatalysts [Figure 2][10]. Specifically, they utilized dialkyl-substituted 2-indolylmethanols 1 as three-atom building blocks to participate in the enantioselective (4 + 3) cycloaddition with dienolsilane 2a under the catalysis of strongly acidic and confined IDPi 4a or 4b. After a subsequent TFA-promoted removal of silyl group, a series of novel bicyclo[3.2.2]cyclohepta[b]indoles 3 bearing three stereogenic centers were synthesized in overall excellent yields with high enantioselectivities. In addition, considering the accessibility of these intriguing bicyclo[3.2.2]cyclohepta[b]indole frameworks, they performed some useful synthetic transformations of products 3 and accomplished the synthesis of enantioenriched indole derivatives 5-8.
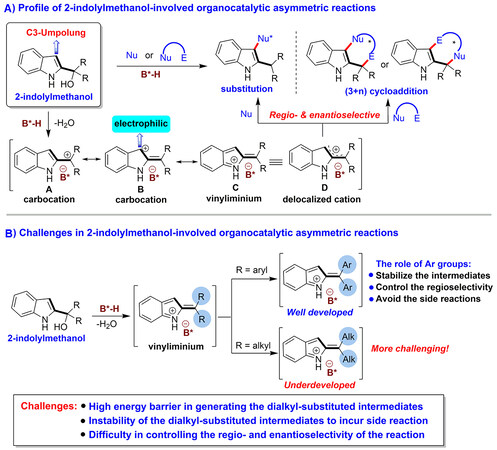
Figure 1. Profile of 2-indolylmethanol-involved organocatalytic asymmetric reactions and the existing challenges.
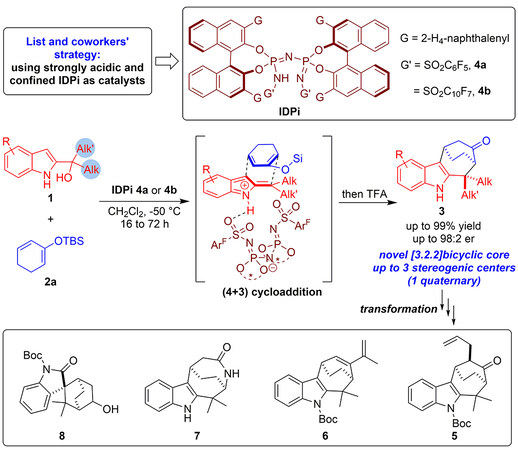
Figure 2. Organocatalytic asymmetric (4 + 3) cycloaddition of dialkyl-substituted 2-indolylmethanols with dienolsilane.
To gain some insights into the reaction mechanism, they performed a series of control experiments and theoretical calculations on this organocatalytic asymmetric (4 + 3) cycloaddition. Based on these investigations, they suggested a possible reaction pathway, which involved an overall concerted yet asynchronous cycloaddition process. As illustrated in Figure 3, initially, strongly acidic and confined IDPi reacted with dienolsilane 2a via a silyl transfer process to generate the active silylium Lewis acid I, which could deliver silyl group to 2-indolylmethanol 1a and form complex II via hydrogen-bonding interaction. Then, complex II underwent the C-O bond cleavage to produce intermediate III along with the release of TBSOH, which was the rate-limiting step with an energy barrier of 14.0 kcal/mol. Subsequently, intermediate III reacted with dienolsilane 2a to undergo a concerted yet highly asynchronous (4 + 3) cycloaddition, which was the enantio-determining step with an energy barrier of 9.6 kcal/mol. The resulted adduct IV underwent a rearomatization process to give intermediate product 3aa’ with the regeneration of IDPi. Finally, the target product 3aa was obtained by TFA-promoted removal of the silyl group in 3aa’.
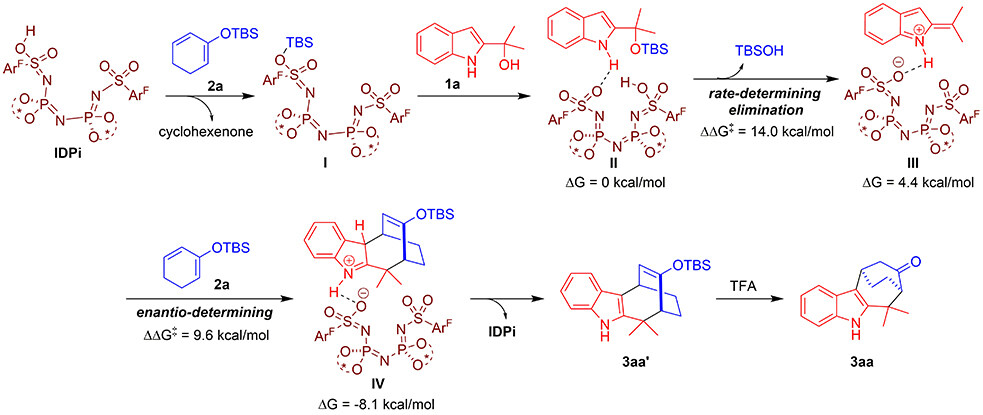
Figure 3. Suggested reaction pathway and activation mode.
In short, List and coworkers established an organocatalytic asymmetric (4 + 3) cycloaddition of dialkyl-substituted 2-indolylmethanols with dienolsilane by using strongly acidic and confined imidodiphosphorimidates as competent organocatalysts, which afforded novel
DECLARATIONS
AcknowledgmentsWe sincerely thank all leading chemists and co-workers involved in the development of 2-indolylmethanol-related chemistry.
Authors’ contributionsWrote the draft manuscript: Tan W
Revised and rewrote some parts of the manuscript: Shi F
Availability of data and materialsNot applicable.
Financial support and sponsorshipWe thank for NSFC (22125104 and 21831007), Natural Science Foundation of Jiangsu Province (BK20210916) and High Education Natural Science Foundation of Jiangsu Province (No. 21KJB150009) for financial supports.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Zhang YC, Jiang F, Shi F. Organocatalytic asymmetric synthesis of indole-based chiral heterocycles: strategies, reactions, and outreach. Acc Chem Res 2020;53:425-46.
2. Mei GJ, Shi F. Indolylmethanols as reactants in catalytic asymmetric reactions. J Org Chem 2017;82:7695-707.
3. Bera K, Schneider C. Brønsted acid catalyzed [3 + 2]-cycloaddition of cyclic enamides with in situ generated 2-methide-2H-indoles: enantioselective synthesis of indolo[1, 2-a]indoles. Org Lett 2016;18:5660-3.
5. Lin X, Wang L, Han Z, Chen Z. Chiral spirocyclic phosphoric acids and their growing applications. Chin J Chem 2021;39:802-24.
6. Sun M, Ma C, Zhou SJ, et al. Catalytic asymmetric (4 + 3) cyclizations of in situ generated
7. Li TZ, Liu SJ, Sun YW, et al. Regio- and enantioselective (3 + 3) cycloaddition of nitrones with 2-indolylmethanols enabled by cooperative organocatalysis. Angew Chem Int Ed Engl 2021;60:2355-63.
8. James T, van Gemmeren M, List B. Development and applications of disulfonimides in enantioselective organocatalysis. Chem Rev 2015;115:9388-409.
9. Gatzenmeier T, van Gemmeren M, Xie Y, Höfler D, Leutzsch M, List B. Asymmetric Lewis acid organocatalysis of the Diels-Alder reaction by a silylated C-H acid. Science 2016;351:949-52.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Tan W, Shi F. A breakthrough in 2-indolylmethanol-involved organocatalytic asymmetric reactions. Chem Synth 2022;2:11. http://dx.doi.org/10.20517/cs.2022.14
AMA Style
Tan W, Shi F. A breakthrough in 2-indolylmethanol-involved organocatalytic asymmetric reactions. Chemical Synthesis. 2022; 2(2): 11. http://dx.doi.org/10.20517/cs.2022.14
Chicago/Turabian Style
Tan, Wei, Feng Shi. 2022. "A breakthrough in 2-indolylmethanol-involved organocatalytic asymmetric reactions" Chemical Synthesis. 2, no.2: 11. http://dx.doi.org/10.20517/cs.2022.14
ACS Style
Tan, W.; Shi F. A breakthrough in 2-indolylmethanol-involved organocatalytic asymmetric reactions. Chem. Synth. 2022, 2, 11. http://dx.doi.org/10.20517/cs.2022.14
About This Article
Copyright

Data & Comments
Data


 Cite This Article 29 clicks
Cite This Article 29 clicks

 Like This Article 8
likes
Like This Article 8
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.