
Molecular chemisorption: a new conceptual paradigm for hydrogen storage


Abstract
Developing efficient hydrogen storage materials and the corresponding methods is the key to successfully realizing the “hydrogen economy”. The ideal hydrogen storage materials should be capable of reversibly ab-/desorbing hydrogen under mild temperatures with high hydrogen capacities. To achieve this target, the ideal enthalpy of adsorption is determined to be 15-50 kJ/mol for hydrogen storage. However, the current mainstream methods, including molecular physisorption and atomic chemisorption, possess either too high or too low enthalpy of hydrogen adsorption, which are not suitable for practical application. To this end, hydrogen storage via molecular chemisorption is perceived to regulate the adsorption enthalpy with intermediate binding energy between the molecular physisorption and atomic chemisorption, enabling the revisable hydrogen ad-/desorption possible under ambient temperatures. In this review, we will elaborate the molecular chemisorption as a new conceptual paradigm and materials design to advance future solid-state hydrogen storage.
Keywords
BACKGROUND
Hydrogen storage plays a critical role in energy transformation from fossil fuels to green energy. To increase the competitiveness of hydrogen as an energy vector, and to unlock its potential as a form of energy storage to smooth intermittent renewable energy production as well as a means of decarbonizing sectors, the storage of hydrogen must be made more efficient, safer, and cheaper, which is essential for future energy security and economic development, whereas it is currently missing in the hydrogen energy supply chain.
Specifically, the hydrogen energy supply chain associated with the “hydrogen economy” is generally composed of hydrogen production, storage/conveyance, and utilization. Up to now, abundant hydrogen sources can be obtained by diverse manufacturing methods, such as hydrocarbon reforming, biomass process, and water splitting. Meanwhile, the mature fuel cell technologies make the commercial application of hydrogen readiness to end-users as well. However, as to hydrogen storage, no practical approach currently exists to fulfill the technical requirements of fuel cells. Firstly, in actual application, hydrogen-powered cars are expected to meet a driving range (> 500 km or 300 miles) on a single fill, which is comparable to conventional combustion cars[1]. This means that about 5-6 kg of hydrogen is required to be stored on-board. Moreover, on-board hydrogen storage is also constrained by safety, weight, efficiency, volume, and cost[2]. So far, despite decades of great efforts, the secure and efficient on-board hydrogen storage method is still the main shackle of converting the world into a “hydrogen economy”[3]. Theoretical innovations and technical breakthroughs are still required to develop secure and efficient hydrogen storage technologies.
DILEMMAS OF CURRENT HYDROGEN STORAGE AND NEW SOLUTION
Currently, the established techniques for hydrogen storage consist of three means: compression, cryogenics, and material-assisted hydrogen storage[4]. Compressing hydrogen into a high-pressure cylinder is a direct and effective method to improve the volumetric energy density of H2. So far, high-pressure cylinders can be pressurized up to 700 bar. However, despite such high pressures, a 150 L storage tank is required to store 6 kg of hydrogen. Moreover, the fast-filling strategies are still immature[5]. Hydrogen storage via liquefaction can offer a higher volumetric density compared with compressed hydrogen storage. Nevertheless, due to the low boiling point of liquid hydrogen, liquefying hydrogen needs to be operated under very low temperatures, which will consume a large amount of energy. Therefore, hydrogen storage systems via cryogenic and high-pressure methods are technically and financially impracticable and nonideal.
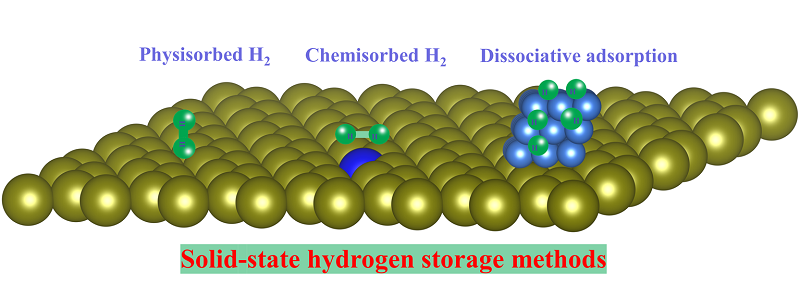
Alternatively, material-assisted solid-state hydrogen storage methods are therefore developed, which can be categorized into physisorption and chemisorption, based on the intrinsic hydrogen adsorption fashions. Hydrogen storage materials via physisorption can achieve higher hydrogen storage capacity on the surface than the bulk concentration due to gas-solid interactions, enabling a high hydrogen storage content[5,6]. Moreover, physiosorbed hydrogen can be readily and reversibly rapid charging/discharging without decomposition of the solid. Basically, materials based on physisorption are dependent on pore size, pore volume, surface area and the hydrogen binding sites decorated in the porous materials. For example,
Blankenship et al.[8] reported cellulose acetate-derived porous carbons which possess high surface area (3800m2·g-1) and pore volume (1.8·cm3·g-1) that arise almost entirely (> 90%) from micropores. The hydrogen storage test indicated a gravimetric hydrogen uptake of 8.1 wt% at -196 °C and 20 bar. However, at room temperature, the carbons only exhibited 1.2 wt% hydrogen uptake at 30 bar. Obviously, in practical, physisorption-type materials are subject to harsh operating conditions (cryogenic -temperature) due to the weak Van der Walls force. As shown in Figure 1, for classical physisorption materials such as the representatives of carbon nanotube, zeolites, covalent organic frameworks, and classical metal-organic frameworks (MOFs)[9-12], their adsorption enthalpies are determined to be 4-10 kJ/mol, which can only stabilize desired hydrogen capacity under the cryogenic temperature. Room temperature will weaken the interaction between H2 molecules and adsorbents, leading to a sharp decrease in hydrogen storage capacity. Stabilizing hydrogen via molecular physisorption at room temperature is still a big challenge. Attempts are also made to enhance the interactions to improve the room temperature stability, such as heteroatom (e.g., O) doped carbon-based materials[8]. The density functional theory study indicates that the doped heteroatoms can modify the electronic structures of substrates via the charge-transfer behavior. This modification to the electronic properties of adsorption sites will create extra dipole moments, thus increasing hydrogen uptake[13]. However, the reported improvement of adsorption enthalpy is limited, generally lower than 15 kJ/mol.
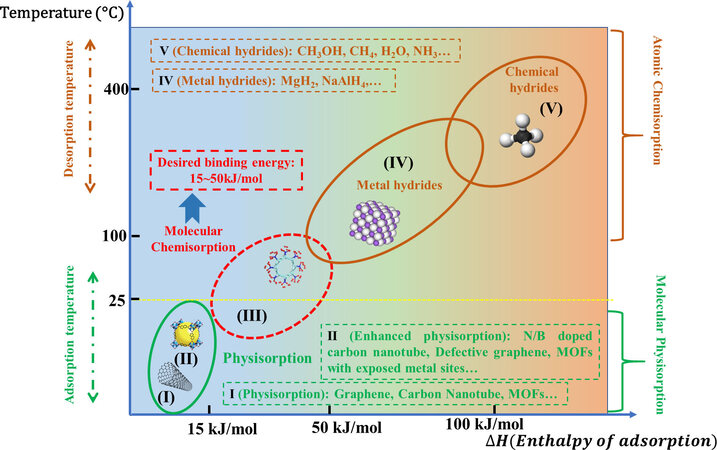
Figure 1. Hydrogen storage methods with respect to enthalpy of adsorption and operating temperature.
In contrast, atomic chemisorption materials, including metal hydrides and chemical hydrides, characteristically possess high thermodynamic stability under room temperature due to the strong bonding between dissociative H atoms and adsorbent atoms. For example, the bonding energy of MgH2 is as large as 75 kJ/mol[14]. Undoutedly, the cleavage of Mg-H requires a high temperature to stimulate the reaction. It is reported that the decomposition of MgH2 is initiated while the temperature is increased up to 300 °C. Such a high operating temperature is not suitable for proton exchange membrane fuel cells[11]. Moreover, MgH2 also suffers from sluggish dynamics. Efforts are also devoted to regulating the thermodynamics of MgH2, such as nanostructuring, alloying, metastable phase[15,16]. However, their hydrogen storage performance still cannot meet the practical demands. For example, Cao et al.[17] reported a Mg85In5Al5Ti5 alloy prepared by plasma milling. Compared with pure MgH2, the measured dehydrogenation enthalpy of Mg85In5Al5Ti5 alloy decreases to 65.2 kJ/(mol·H2). However, kinetic properties indicate that a high temperature (over 300 °C) is still required to achieve rapid dehydrogenation kinetics. Chemical hydrides, like NaBH4 and NH3BH3, generally possess higher hydrogen capacity[18,19]. However, their drawbacks are severe. Taking NaBH4 as an example, hydrogen release by the thermolysis of NaBH4 is impractical due to the high thermodynamic stability, stepwise decomposition companied with impurity gases and poor dynamics. It reported that the decomposition temperature of NaBH4 was determined to be higher than 500 °C[19-21]. Alternatively, employing hydrolysis as off-board hydrogen storage, NaBH4 still suffers from non-recyclability and non-affordability issues, despite its high hydrogen capacity and controllable hydrogen release. According to the guidelines set by the United States Department of Energy (DOE) for onboard hydrogen storage, by the year 2025, the gravimetric and volumetric storage capacities should meet the target of 5.5 wt% and 40 g/L under near-ambient operation temperatures of -40 to 85 °C[22].
Obviously, neither molecular physisorption nor atomic chemisorption could meet the targets of DOE. The inherent issues of the two hydrogen storage methods are ascribed to the unsuitable adsorption enthalpies of hydrogen. The reported solutions are mainly focused on how to improve weak molecular physisorption, or to decrease the strong atomic chemisorption, so as to form moderate binding energy. However, none of the current materials can meet the practical application requirements. The ideal hydrogen storage materials are reversibly operated under mild temperatures with fast charging/discharging kinetics. In addition, according to the DOE target, the hydrogen storage materials should be capable of adsorbing at least 5.5 wt% of hydrogen. This means that hydrogen can be stored in molecular form, but in a weak chemisorption manner, called molecular chemisorption. This fascinating hydrogen storage method simultaneously endows materials the good reversibility as physical adsorption and moderate stability under room temperature as chemical adsorption, which may stand out as a promising hydrogen storage method.
THE MOLECULAR-CHEMISORPTION of HYDROGEN
The molecular chemisorption process can be classified into weak chemisorption involving the gas-solid interactions on the surface. Hence, the structure of adsorption sites at the surface is vital to uncovering the underlying mechanism of molecular chemisorption, which is the prerequisite to both materials design and hydrogen adsorption behavior. Early in 1984, Kubas et al.[23] experimentally discovered a non-dissociative form of hydrogen adsorbed onto a coordinated transition metal (TM) complex, W(CO)3(P/Pr3)2. In this TM complex, the W atom is linked with five coordinates, and the hydrogen molecule is chemisorbed without dissociation via side-on coordination binding to the W atom. However, the bond length of hydrogen is stretched by approximately 20% longer than that of free H2. To explain the dihydrogen binding coordination, the Dewar-Chatt-Duncanson model for TM-olefin complexes[24] is invoked to rationalize the stability of these σ-type complexes, called Kubas-type interaction. Similar to the Dewar-Chatt-Duncanson model with a three-center two-electron (3c-2e) bond (type I shown in Figure 2), the Kubas-type interaction involves σ donation from the filled σ-bonding orbital of H2 molecule to an unfilled d orbital of a TM. Meanwhile, π back-donation (BD) occurs from a filled d orbital of a TM to the empty σ* anti-bonding orbital of the H2 molecule, which determines the stability of molecularly chemisorbed H2 (type II shown in Figure 2).
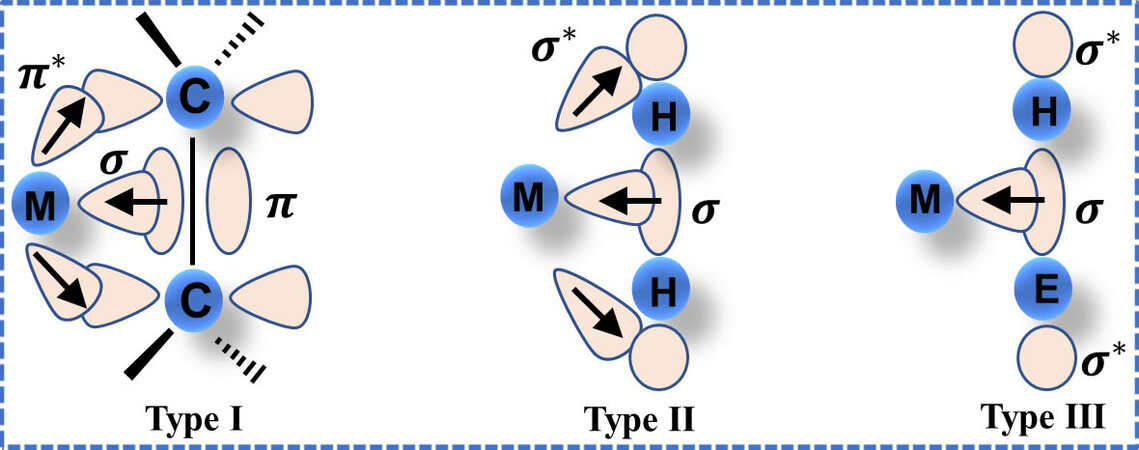
Figure 2. Schematic diagram showing three orbital interactions between the central element and ligand. Type I: TM-olefin complexes. Type II: Kubas-type H2 complexes. Type III: η2-σ complexes (E represents H, C, Si, etc.).
The non-dissociative molecular chemisorption has ever been mentioned in metal-based hydrogen storage materials. It is known that the hydrogen sorption process upon metal atoms includes: physisorption, dissociation, hydrogen atom diffusion, hydride nucleation and growth. Note that the physisorbed H2 will enter a molecular chemisorption state after crossing an activation barrier. The molecular chemisorption is generally considered a transition state before the dissociation of hydrogen. Mårtensson et al.[25] observed the molecular H2 chemisorbed at the edge sites of a stepped Ni (100) surface that is covered with a dense layer of atomic hydrogen by use of low-energy electron diffraction and electron energy-loss spectroscopy (EELS) in conjunction. In contrast, no such state is observed on the flat Ni (100) surface. The authors ascribed the molecular chemisorption to Kubas-type interaction. They suggested that nickel atoms with lower coordination at the stepped surface possess residual unfilled d states which could stabilize the chemisorbed H2, while the dissociative channel is blocked by the saturated atomic adsorbate layer. The results illustrated that the low-coordinated metal atoms possess the capacity of molecular chemisorption.
Recently, Berwanger et al.[26] reported the direct observation of chemical binding forces on single Fe atoms to study the chemical reactivity of atom-assembled Fe clusters from 1 to 15 atoms at an atomic scale. This measurement can be achieved via scanning probe microscopy with CO-terminated tips, which can transition from the physisorption to the chemisorption regime and form a weak chemical bond with the individual atoms. It is found that the chemisorption strength increases inversely with the atom’s reduced coordination number. The results imply that regulating atomic coordination numbers can enhance the interaction force between H2 and adsorbent atoms.
The above analysis indicates that the low-coordinated metal atoms possess the capacity of molecular chemisorption. However, this does not mean that molecular chemisorption is Kubas action. According to Kubas interaction, it is the TM atom (owning d-orbital) with an unsaturated coordination state that can chemisorb H2 in its molecular form. To some extent, Kubas-type interaction is reasonable, which explains the molecular chemisorption of H2 and its stability, especially for TM atoms. However, the Kubas-type interaction is not a versatile theory or not the unique premise for molecular chemisorption in some cases. The first case is the MOFs with exposed metal sites (coordination-unsaturated). Naturally, MOFs with exposed metal sites possess uniformly distributed metal sites coordinated to organic linkers and sufficient porosity. It is reported that MOFs with exposed metal sites can significantly enhance the enthalpies of adsorption of H2 compared to classical MOFs with saturated metal sites. For example, the FT-IR spectrometry studies indicate that H2 can be adsorbed onto the coordinatively-unsaturated nickel (II) sites of MOF-74 with a strong adsorption enthalpy of 13.5 kJ·mol-1[27]. In contrast, this elevated interaction of TM-H2 is not found in classical MOFs with saturated TMs. However, the hydrogen adsorption mechanism for MOFs with exposed metal sites still involves physisorption that the hydrogen capacities are mostly rely on the surface area and total pore volume of the material, despite improved enthalpy of hydrogen adsorption. Zhou and Yildirim[28] individually investigate the role of exposed TM sites in MOFs by first-principles calculations as well. Characteristics including charge transfer from TM to H2, H2-σ* Mn-d orbital hybridization, and H-H bond elongation, are not observed in the H2-MOFs (with exposed TM sites) system, indicating that the enhanced heat of adsorption of H2 is not related to the Kubas-type interaction. Furthermore, the authors reveal that the improved binding energy is contributed by the classical Coulomb interaction arising from the small charge overlap of H2-σ and TM-d orbitals.
Another example is the adsorbent containing metals without d-electrons. As detailed in Kubas-type interaction, the adsorption site of transition metal should possess at least a vacant d orbit to accept the electrons from H2 molecule as well as a filled d orbit to back-donate electrons to the anti-bonding orbit of H2 molecule. Here, one may wonder whether they are capable of adsorbing intact H2 molecules while the adsorption sites contain d0 metals or high-oxidation-state main group elements that lack of electrons in the d orbital? According to Kubas, if there is a lack of BD, the Kubas-type H2 complexes will be unstable. However, in recent research, Wang et al. indicate that metals lacking d orbital electrons still stably adsorb intact H2 molecules, referred to as η2-σ complexes in type-III [Figure 2][29]. These findings illustrate that supported alkali metals or alkaline earth metals (absence of d-electron), like Li- or Ca-modified carbon nanotube, possess the potential to be utilized for hydrogen storage. The results further indicate that the BD is not the crucial factor for the stabilization of molecularly chemisorbed H2.
Furthermore, a strong polarization effect of low-valent TM on hydrogen adsorption is also significant. Based on self-consistent-field molecular-orbital theory, Niu et al.[30] reported that hydrogen behaves differently while approaching a neutral metal atom (Ni) and a metal ion (Ni+). For a neutral metal atom, similar to the metal atoms on surfaces and in bulk, it will transfer an electron to the approaching H2 molecule to fill the antibonding orbital. This will lead to the bond cleavage of H-H favoring the atomic bonding between individual H atoms and metal atoms. In contrast, the ability of electron donation to a H2 molecule becomes energetically inefficient for a transition metal ion due to the higher second ionization potential of the metal atom. Instead, a strong polarization effect will occur between the ion and H2 molecule, which will enable several H2 molecules to be chemisorbed in their molecular form onto a metal ion.
However, an isolated neutral metal atom or low-valent metal ion is unstable and cannot exist alone in nature. In addition, in a practical hydrogen storage material containing low-valent metal ions, the interactions between metal sites and H2 molecules may involve multiple contributions to molecular chemisorption. As to the interactions mentioned above, combined with the Kubas-type interaction, it can be concluded that it is the coordinated TM atom, not a neutral metallic atom, that can bind H2 in the form of a molecule. Moreover, the low-valent TM atoms with unsaturated configuration are vital for molecular chemisorption via orbital interaction, rather than physisorption.
Besides the low-valent and unsaturation state of TM atoms, the local electronic structure of TM atoms also contributes to adsorption energy. Recently, Zhang et al.[31] reported a low-coordinate Pt1 single-atom catalyst for the hydrogenation of nitrobenzene and cyclohexanone. By impregnation-reduction steps, isolated Pt atoms can be stabilized on phosphomolybdic acid (PMA)-modified activated carbon (AC) (Pt-PMA/AC). To investigate the catalytic activity of Pt-PMA/AC in hydrogenation reactions, the authors firstly conducted a H2-O2 titration experiment [Figure 3A-C]. It is found that H2 uptake of Pt-PMA/AC is due to the presence of Pt. In contrast, neither AC nor PMA/AC could adsorb H2. This explained that the anchored Pt atoms strongly interact with PMA, inducing an electron out-flow. Thus, the Pt single atom enables the Pt-PMA/AC to possess the excellent hydrogen uptake property by its strong adsorption and polarization capacity. Besides the single atom, dual-metal dimers are also found to be capable of strong adsorption capacity. Zhang et al.[32] reported an asymmetrically heterogeneous atomic metal (Co and Pt) on N8V4 vacancies by the defect engineering method, shown in Figure 3D and E. The asymmetric deployment of Pt and Co in a(Co-Pt)@N8V4 can polarize the surface charges near the active sites by stimulating the electron accumulation around the Co atom and weakening electron accumulation around Pt [Figure 3F]. This charge distribution significantly enhances the adsorption of adsorbate molecules on active sites. Moreover, the asymmetric coordination of heterobimetallic species coupling at carbon defects can shift up the d orbital relative to the Fermi level, improving the binding energy between adsorbate and adsorption sites.
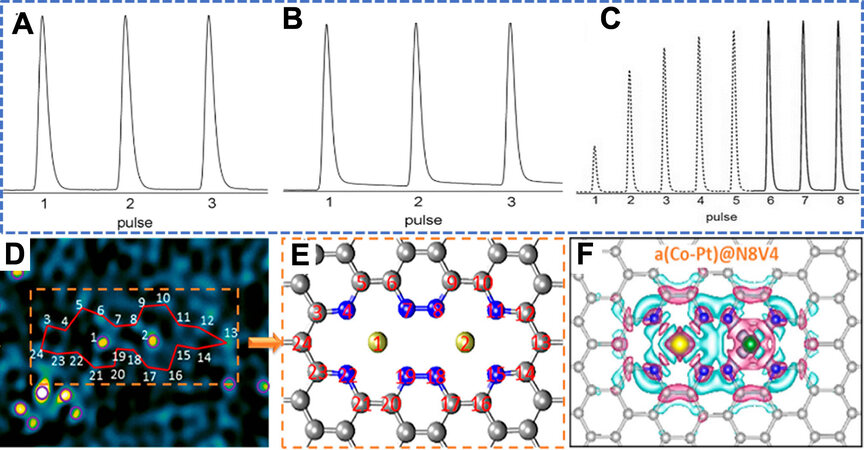
Figure 3. The atomically dispersed asymmetrical metallic structure. (A-C) H2-O2 titration profiles of AC, PMA/AC and Pt-PMA/AC. Reprinted with permission from Ref.[31]. Copyright 2016, WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim. (D) HAADF image of A-CoPt-NC after fast Fourier transformation filtering. The bright yellow spots are metal atoms, and the cyan spots are carbon atoms. Reprinted with permission from Ref.[32]. Copyright 2018, American Chemical Society. (E) Model of the configuration of the two metal atoms trapped in the defect, reconstructed from the observed atomic structure in (a). Purple circles mark metal atoms. The carbon atoms adjacent to metal atoms #1 and #2 are linked with a red line. Reprinted with permission from Ref.[32]. Copyright 2018, American Chemical Society. (F) Top view of the charge densities of a(Co-Pt)@N8V4, the pink area represents strong electron accumulation. Reprinted with permission from Ref.[32]. Copyright 2018, American Chemical Society.
Based on the above analysis, a low valent ion state for coordinated TM atoms is the prerequisite of hydrogen adsorption without dissociation. Besides, the stabilization of chemisorbed molecular hydrogen under room temperature might need further modulation of the electronic structures to adsorption sites. On the one hand, the adsorption sites should possess a low coordination number. According to the d band center theory proposed by Hammer et al.[33], the lowering coordination number of center metal can shift the d-band level up to the Fermi level, which could strengthen the interaction between adsorption sites and adsorbate molecules. On the other hand, the asymmetric adsorption sites can effectively alter the charge distribution at the coordination structures, which will lead to local charge accumulation. This kind of charge redistribution could further promote the adsorption of adsorbate molecular by strong molecular deformation.
Previous experimental explorations further confirmed the role of the unique material structure on molecular chemisorption for hydrogen storage. Strategies of material synthesis include Grafting TM atoms to surfaces of porous materials (e.g., mesoporous silica)[34,35] and bridging the TM atoms with lightweight ligands (e.g., hydrazine) to the microporous materials[36-38]. In 2008, Hamaed et al.[34] reported organometallic Ti fragments as the H2 storage material. The low-coordinated Ti (III) atoms were grafted onto mesoporous silica using tri- and tetrabenzyl Ti precursors. The detailed experimental process is shown in Figure 4A. It can be observed that the low valent Ti atoms are three coordinates with two O atoms and one C atom, which can act as the adsorption sites. The hydrogen storage properties in Figure 4B show that the highest gravimetric storage value of 2.02 wt% recorded under the condition of 60 atm and 77 K is the sample with a Ti/Si molar ratio of 0.2 (0.20 Ti/HMS), while only 1.21 wt% hydrogen can be stored by silica precursor under the same condition. By calculations, an average of 2.7 H2 molecules are adsorbed onto each Ti atom in 0.20 Ti/HMS. Notably that hydrogen adsorption isotherms of Ti-grafted samples do not saturate under the condition of 60 atm and 77 K. Increasing the pressure can further increase the numbers of H2 molecules being adsorbed, according to the Le Chatelier’s principle. Moreover, the binding enthalpy for Ti (III) grafted sample, by Clausius-Clapyron equation, is determined to be 22.04 kJ/mol compared to 3.5 kJ/mol for the pristine silica materials. On the one hand, the dramatically enhanced enthalpy reflects the molecular chemisorption mechanism differing from typical physical adsorption characteristics. On the other hand, the large enthalpy guarantees that around 1.1 hydrogen molecules are molecularly chemisorbed onto each Ti atom at room temperature.
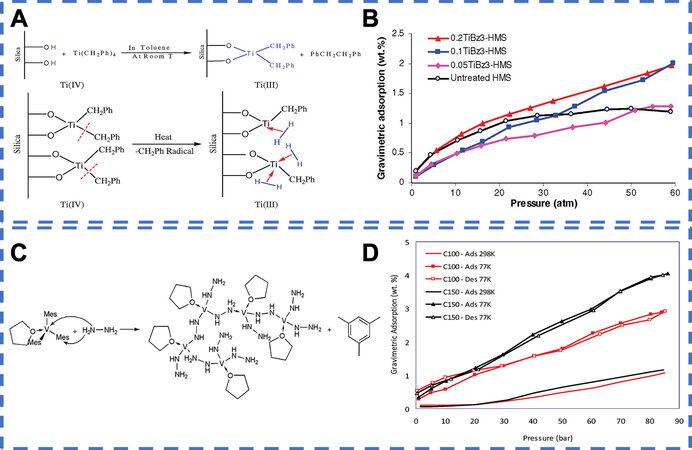
Figure 4. Hydrogen storage performance (via molecular chemisorption). (A) Schematic representation of the grafting of benzyl Ti species onto the surface of mesoporous silica. Reprinted with permission from Ref.[34]. Copyright 2008, American Chemical Society. (B) Hydrogen adsorption isotherms at 77 K for mesoporous silica treated with various molar equivalents of tribenzyl titanium. Reprinted with permission from Ref.[34]. Copyright 2008, American Chemical Society. (C) A possible mechanism for the reaction between V(Mes)3·THF and N2H4. Reprinted with permission from Ref.[38]. Copyright 2010, American Chemical Society. (D) Hydrogen adsorption-desorption isotherms of vanadium hydrazide samples. Reprinted with permission from Ref.[38]. Copyright 2010, American Chemical Society.
In addition to grafting techniques, vanadium hydrazide gels with isolated and coordination-unsaturated V (III) species are also fabricated by using trismesitylvanadium (III) as a precursor and hydrazine as a linker[38]. The possible reaction mechanism is given in Figure 4C. The vanadium atom is tri-coordinated with N atoms in vanadium hydrazide gels, though some residual tetrahydrofuran (THF) molecules are bound to V atoms. The hydrogen adsorption isotherms [Figure 4D] present a linear hydrogen adsorption behavior, reflecting nondissociative hydrogen chemisorption, and up to 4.04 wt% of hydrogen are stored for sample C150 under 77 K and 85 bar. Even at room temperature, 49% of their adsorption can be retained as opposed to 77 K, owing to its greatly enhanced adsorption enthalpies from 3 to 36.5 kJ/mol.
MATERIAL DESIGN
Note that, though the works mentioned above exhibited good hydrogen adsorption performance, the lack of details of material structure characterization is unfavorable to our cognition of constitutive relationship, and it also brings difficulties to the further exploration of related materials. The core of rationalizing materials capable of molecular chemisorption is to fabricate isolated heteroatoms, simultaneously with low coordination number and unique coordination configuration. The expectation is that, by regulating the electronic structures of adsorption sites, the H2 molecules can be chemisorbed with moderate bond energy, in which H2 molecules can be adsorbed stably and released readily while exposed to a vacuum. Nevertheless, exploration and development of synthetic methodologies is a great challenge because it is difficult to synthesize a material that simultaneously has a high concentration of controllable coordination structures with unsaturated center metal atoms, lightweight and sufficient porosity, allowing the diffusion of hydrogen[39]. On the one hand, the high surface energy easily prompts the migration and aggregation of isolated metal atoms into particles. On the other hand, TMs in the solid state prefer to be hexa-coordinate in most cases, such as TiO2, hindering the hydrogen molecular chemisorption. Experimental literature related to molecular chemisorption is rarely reported. Here, the methodology, defect engineering strategy, is highlighted as a simple and versatile strategy that could not only fabricate atomically dispersed and low-coordinate transition atoms but also modulate the coordination structure of centred TM atoms.
Defective carbon-based materials are widely considered as the ideal substrates owing to their good structural tunability, sufficient porosity, and lightweight. More importantly, the abundant defects in the carbon matrix could act as the anchoring sites to stabilize isolated TM atoms at atomic levels. Experiments have identified that a di-vacancy defect in graphene can trap atomic Ni specie in form of a four-coordinate structure (Ni-C4) which exhibits excellent stability, shown in Figure 5A[40]. Moreover, the defect engineering strategy can be employed to regulate TM-atom-coordinated structures with different non-metallic element atoms (B, N and/or S). For example, the regulation of coordination configuration of defect-trapped transition metal atoms can be achieved by selectively replacing C atoms at defects with N atoms, shown in Figure 5B[41]. Furthermore, lowering coordination numbers by directly removing partial ligands seems feasible via an Ar-plasma etching technique[42]. Besides the configuration of the di-vacancy defect trapping a single Ni, the defect engineering can be applied to synthesize configurations that heterogeneous di-metal or even tri-metal confined into a multi-vacancy defect for hydrogen storage[43].
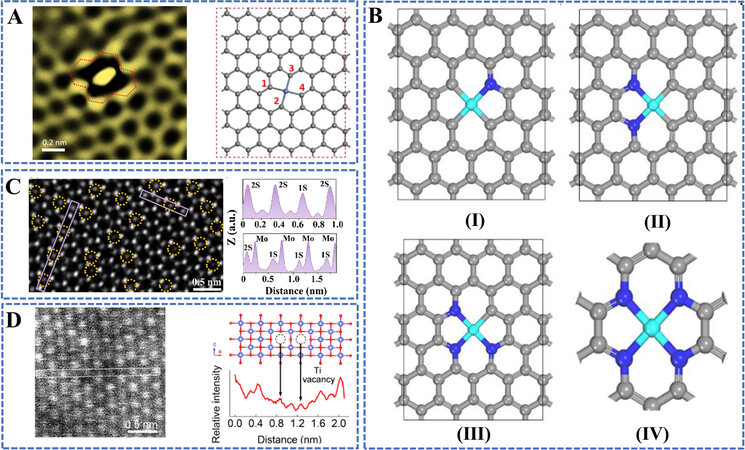
Figure 5. Methodology of the defect engineering. (A) HAADF-STEM image of the defective area with atomic Ni trapped (left) and the corresponding illustration of a single Ni atom anchored on di-vacancy (right). Reprinted with permission from Ref.[40]. Copyright 2018, Elsevier. (B) Top views of the optimized structures for CoN4-xCx. (I-IV) Represent four different coordination environments of cobalt atoms; I: CoN1C3, II: CoN2C2, III: CoN3C1, IV: CoN4. Reproduced with permission from Ref.[41]. Copyright 2020, Wiley-VCH. (C) STEM image together with the line profiles extracted from the areas marked with purple rectangles of a monolayer MoS2 flake film after etching. Reprinted with permission from Ref.[45]. Copyright 2020, American Chemical Society. (D) HAADF STEM images of a
In addition to carbon-based materials, the defect engineering strategy can also be applied to transition metal chalcogenides by creating ion vacancy defects, which could not only modulate the coordination structure and charge distribution of center TM atoms but also optimize steric effect of H2 adsorption. Reducing the coordination number of TM atoms by engineering anionic vacancy defects is practicable. Experimentally, the Ar-Plasma technique has successfully witnessed the removal of partial coordinated anionic ligands in transition metal chalcogenide[44]. Besides, chemical etching is also reported to create homogeneously distributed single S-vacancy defects onto the MoS2 nanosheet surface with a high vacancy concentration of 12.11%, shown in Figure 5C[45].
Besides anionic vacancy defects, engineering cationic vacancy defects is also crucial for binding hydrogen via molecular chemisorption. The creation of cationic vacancies can undoubtedly enlarge the adjacent TM-TM distance, thus influencing the hydrogen adsorption behavior. Various techniques have been developed to create cationic vacancies, such as strong alkali etching[46], wet-chemistry approach[47], high-intensity ultrasonic processing[48] and so on. In Figure 5D, the HAADF-STEM image and line profile confirmed the Ti vacancy defects in the cation-deficient titanium oxide (Ti0.87O2) fabricated by alkali etching[46]. In addition to producing ion vacancy defects, the coordination environment and charge distribution of center metals can also be modified by introducing heteroatoms (metal or non-metal atoms) into the defects[44,49].
PERSPECTIVES AND DIRECTIONS
Developing hydrogen storage materials in the form of molecular chemisorption will construct a new conceptual paradigm in solid-state hydrogen storage fields. This unique hydrogen storage fashion endows the materials with fascinating adsorption enthalpy, allowing the hydrogen adsorption under ambient temperature and fast dehydrogenation simply by exposure to a vacuum condition. Though it is still premature to be fully understood, this method will provide a potential solution to hydrogen storage in the future. Currently, it remains a big challenge for current fabrication processes. The aforementioned defect engineering offers a doable way, albeit it needs to be further verified for hydrogen storage. Efforts to explore the unique hydrogen storage materials and understand the adsorption mechanism is urgently required.
1. Strategies of synthesizing molecular-chemisorption type materials aim to acquire high concentration of low-valent, low-coordinate binding sites.
2. Factors that affect the electronic structures of adsorption sites should be delicately and sophisticatedly modulated, including: (1) adsorption sites, such as species, spacing and quantity of adsorbent atoms; (2) coordination environment, such as coordination number, coordination atom, symmetry/asymmetry of coordination structure, and position (in plane or edge); and (3) substrates.
3. The cycle stability of materials during de-/hydrogenation is a non-ignorable performance. The interaction between ligands and central TM atoms should be fully considered prior to material design.
4. More accurate theoretical calculations combined with matching adsorption models should be provided to explore the critical interactions behind the molecular chemisorption, favoring the correlation between structure of adsorption sites with hydrogen adsorption performance.
5. Besides the TM atoms, efforts should be also devoted into developing η2-σ complexes (type III method referred in Figure 2) constructing an integrated molecular chemisorption system.
DECLARATIONS
Authors’ contributionsConceived and wrote the paper: Lang C, Jia Y, Yan X, Ouyang L, Zhu M, Yao X
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work has been supported by the Australian Research Council (ARC DP200103043) and ARC Discovery Early Career Researcher Award (ARC DE180101030).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Weidenthaler C, Felderhoff M. Solid-state hydrogen storage for mobile applications: Quo Vadis? Energy Environ Sci 2011;4:2495.
3. Lang C, Jia Y, Yao X. Recent advances in liquid-phase chemical hydrogen storage. Energy Stor Mater 2020;26:290-312.
4. Schlapbach L and Züttel A. Hydrogen-storage materials for mobile applications. Nature 2001;414:353-8.
5. Dalebrook AF, Gan W, Grasemann M, Moret S, Laurenczy G. Hydrogen storage: beyond conventional methods. Chem Commun (Camb) 2013;49:8735-51.
6. Chen Z, Kirlikovali KO, Idrees KB, Wasson MC, Farha OK. Porous materials for hydrogen storage. Chem 2022; doi: 10.1016/j.chempr.2022.01.012.
7. Li Y, Xiao Y, Dong H, Zheng M, Liu Y. Polyacrylonitrile-based highly porous carbon materials for exceptional hydrogen storage. Int J Hydrogen Energy 2019;44:23210-5.
8. Blankenship LS, Balahmar N, Mokaya R. Oxygen-rich microporous carbons with exceptional hydrogen storage capacity. Nat Commun 2017;8:1545.
10. Sun G, Tangpanitanon J, Shen H, et al. Physisorption of molecular hydrogen on carbon nanotube with vacant defects. J Chem Phys 2014;140:204712.
11. Panchariya DK, Rai RK, Anil Kumar E, Singh SK. Core-shell zeolitic imidazolate frameworks for enhanced hydrogen storage. ACS Omega 2018;3:167-75.
12. Lee S, Lee J, Kim Y, Kim J, Lee K, Park S. Recent progress using solid-state materials for hydrogen storage: a short review. Processes 2022;10:304.
13. Shayeganfar F, Shahsavari R. Oxygen- and lithium-doped hybrid boron-nitride/carbon networks for hydrogen storage. Langmuir 2016;32:13313-21.
14. Yu H, Bennici S, Auroux A. Hydrogen storage and release: kinetic and thermodynamic studies of MgH2 activated by transition metal nanoparticles. Int J Hydrogen Energy 2014;39:11633-41.
15. Zhang J, Li Z, Wu Y, et al. Recent advances on the thermal destabilization of Mg-based hydrogen storage materials. RSC Adv 2019;9:408-28.
16. Luo Q, Li J, Li B, Liu B, Shao H, Li Q. Kinetics in Mg-based hydrogen storage materials: enhancement and mechanism. J Magnes Alloy 2019;7:58-71.
17. Cao Z, Ouyang L, Wu Y, et al. Dual-tuning effects of In, Al, and Ti on the thermodynamics and kinetics of Mg85In5Al5Ti5 alloy synthesized by plasma milling. J Alloys Compd 2015;623:354-8.
18. Konarova M, Tanksale A, Norberto Beltramini J, Qing Lu G. Effects of nano-confinement on the hydrogen desorption properties of MgH2. Nano Energy 2013;2:98-104.
19. Martelli P, Caputo R, Remhof A, Mauron P, Borgschulte A, Züttel A. Stability and decomposition of NaBH4. J Phys Chem C 2010;114:7173-7.
20. Urgnani J, Torres F, Palumbo M, Baricco M. Hydrogen release from solid state NaBH4. Int J Hydrogen Energy 2008;33:3111-5.
21. Mao J, Yu X, Guo Z, Liu H, Wu Z, Ni J. Enhanced hydrogen storage performances of NaBH4-MgH2 system. J Alloys Compd 2009;479:619-23.
22. García-holley P, Schweitzer B, Islamoglu T, et al. Benchmark study of hydrogen storage in metal-organic frameworks under temperature and pressure swing conditions. ACS Energy Lett 2018;3:748-54.
23. Kubas GJ, Ryan RR, Swanson BI, Vergamini PJ, Wasserman HJ. Characterization of the first examples of isolable molecular hydrogen complexes, M(CO)3(PR3)2(H2) (M = molybdenum or tungsten; R = Cy or isopropyl). Evidence for a side-on bonded dihydrogen ligand. J Am Chem Soc 1984;106:451-2.
25. Mrtensson A, Nyberg C, Andersson S. Observation of molecular H2 chemisorption on a nickel surface. Phys Rev Lett 1986;57:2045-8.
26. Berwanger J, Polesya S, Mankovsky S, Ebert H, Giessibl FJ. Atomically resolved chemical reactivity of small Fe clusters. Phys Rev Lett 2020;124:096001.
27. Vitillo JG, Regli L, Chavan S, et al. Role of exposed metal sites in hydrogen storage in MOFs. J Am Chem Soc 2008;130:8386-96.
28. Zhou W, Yildirim T. Nature and tunability of enhanced hydrogen binding in metal-organic frameworks with exposed transition metal sites. J Phys Chem C 2008;112:8132-5.
29. Liu Y, Su B, Dong W, Li ZH, Wang H. Structural characterization of a boron(III) η2-σ-silane-complex. J Am Chem Soc 2019;141:8358-63.
30. Niu J, Rao BK, Jena P. Binding of hydrogen molecules by a transition-metal ion. Phys Rev Lett 1992;68:2277-80.
31. Zhang B, Asakura H, Zhang J, Zhang J, De S, Yan N. Stabilizing a Platinum1 single-atom catalyst on supported phosphomolybdic acid without compromising hydrogenation activity. Angew Chem 2016;128:8459-63.
32. Zhang L, Fischer JMTA, Jia Y, et al. Coordination of atomic Co-Pt coupling species at carbon defects as active sites for oxygen reduction reaction. J Am Chem Soc 2018;140:10757-63.
33. Hammer B, Nørskov J. Theoretical surface science and catalysis-calculations and concepts. Impact of Surface Science on Catalysis. Elsevier; 2000. pp. 71-129.
34. Hamaed A, Trudeau M, Antonelli DM. H2 storage materials (22 KJ/mol) using organometallic Ti fragments as sigma-H2 binding sites. J Am Chem Soc 2008;130:6992-9.
35. Hoang TKA, Hamaed A, Trudeau M, Antonelli DM. Bis(benzene) and Bis(cyclopentadienyl) V and Cr doped mesoporous silica with high enthalpies of hydrogen adsorption. J Phys Chem C 2009;113:17240-6.
36. Xu G, Pareek K, Li N, Cheng H. High capacity hydrogen storage at room temperature via physisorption in a coordinatively unsaturated iron complex. Int J Hydrogen Energy 2015;40:16330-7.
37. Hoang TK, Hamaed A, Moula G, Aroca R, Trudeau M, Antonelli DM. Kubas-type hydrogen storage in V(III) polymers using tri- and tetradentate bridging ligands. J Am Chem Soc 2011;133:4955-64.
38. Hoang TK, Webb MI, Mai HV, et al. Design and synthesis of vanadium hydrazide gels for Kubas-type hydrogen adsorption: a new class of hydrogen storage materials. J Am Chem Soc 2010;132:11792-8.
39. Skipper CVJ, Antonelli DM, Kaltsoyannis N. Are metal-metal interactions involved in the rising enthalpies observed in the Kubas binding of H2 to hydrazine-linked hydrogen storage materials? J Phys Chem C 2012;116:19134-44.
40. Zhang L, Jia Y, Gao G, et al. Graphene defects trap atomic Ni species for hydrogen and oxygen evolution reactions. Chem 2018;4:285-97.
41. Yang Q, Jia Y, Wei F, et al. Understanding the Activity of Co-N4-xCx in atomic metal catalysts for oxygen reduction catalysis. Angew Chem 2020;132:6178-83.
42. Tao L, Wang Q, Dou S, et al. Edge-rich and dopant-free graphene as a highly efficient metal-free electrocatalyst for the oxygen reduction reaction. Chem Commun (Camb) 2016;52:2764-7.
43. Zhu X, Zhang D, Chen C, et al. Harnessing the interplay of Fe-Ni atom pairs embedded in nitrogen-doped carbon for bifunctional oxygen electrocatalysis. Nano Energy 2020;71:104597.
44. Zhuang L, Jia Y, Liu H, et al. Defect-Induced Pt-Co-Se coordinated sites with highly asymmetrical electronic distribution for boosting oxygen-involving electrocatalysis. Adv Mater 2019;31:e1805581.
45. Wang X, Zhang Y, Si H, et al. Single-atom vacancy defect to trigger high-efficiency hydrogen evolution of MoS2. J Am Chem Soc 2020;142:4298-308.
46. Xiong P, Zhang X, Zhang F, et al. Two-dimensional unilamellar cation-deficient metal oxide nanosheet superlattices for high-rate sodium ion energy storage. ACS Nano 2018;12:12337-46.
47. Liu B, Wang Y, Peng HQ, et al. Iron vacancies induced bifunctionality in ultrathin feroxyhyte nanosheets for overall water splitting. Adv Mater 2018:e1803144.
48. Dou Y, He CT, Zhang L, et al. Approaching the activity limit of CoSe2 for oxygen evolution via Fe doping and Co vacancy. Nat Commun 2020;11:1664.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lang C, Jia Y, Yan X, Ouyang L, Zhu M, Yao X. Molecular chemisorption: a new conceptual paradigm for hydrogen storage. Chem Synth 2022;2:1. http://dx.doi.org/10.20517/cs.2021.15
AMA Style
Lang C, Jia Y, Yan X, Ouyang L, Zhu M, Yao X. Molecular chemisorption: a new conceptual paradigm for hydrogen storage. Chemical Synthesis. 2022; 2(1): 1. http://dx.doi.org/10.20517/cs.2021.15
Chicago/Turabian Style
Lang, Chengguang, Yi Jia, Xuecheng Yan, Liuzhang Ouyang, Min Zhu, Xiangdong Yao. 2022. "Molecular chemisorption: a new conceptual paradigm for hydrogen storage" Chemical Synthesis. 2, no.1: 1. http://dx.doi.org/10.20517/cs.2021.15
ACS Style
Lang, C.; Jia Y.; Yan X.; Ouyang L.; Zhu M.; Yao X. Molecular chemisorption: a new conceptual paradigm for hydrogen storage. Chem. Synth. 2022, 2, 1. http://dx.doi.org/10.20517/cs.2021.15
About This Article
Copyright

Data & Comments
Data


 Cite This Article 33 clicks
Cite This Article 33 clicks

 Like This Article 26
likes
Like This Article 26
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.