
Advances in radical phosphorylation from 2016 to 2021


Abstract
Organophosphorus compounds are of great significance in organic chemistry. Therefore, the construction of phosphorus-containing compounds has attracted significant attention from organic chemists. Radical phosphorylation has become a powerful strategy for building organophosphorus compounds and numerous achievements have been realized in recent years. In this review, we summarize the development of the generation and application of phosphorus radicals in organic chemistry since 2016. Special emphasis is given to various new transformations involving the generation of P-centered radicals via transition metal catalytic, photochemical and electrochemical means. Recent advances in the development of metal-free catalytic phosphorylations involving P-centered radicals are also reviewed.
Keywords
INTRODUCTION
Organophosphorus compounds are highly important and have been widely applied in the fields of materials science, medicinal chemistry and organic synthesis[1-3]. The introduction of phosphorus functionalities into molecules can be used to effectively modify their medicinal properties and biological responses. Traditional strategies for the formation of C-P bonds heavily rely on nucleophilic substitution, such as the Arbuzov reaction, Friedel-Crafts-type reaction of PCl3 with aromatic compounds and the reactions of organometallic reagents with electrophilic phosphorus species, including Ph2P(O)Cl[4]. Although the transition metal-catalyzed cross-coupling reactions of C-X bonds between organo-(pseudo)halides with P(O)-H compounds have been demonstrated to be a powerful method for C-P bond formation[5], the development of other efficient methods from inexpensive and readily available starting materials for the construction of new organophosphorus compounds remains highly important and has thus gained significant attention from the organic chemistry community.
In recent decades, along with the rapid development of radical chemistry, the construction of X-P (X=C, N, O or S) bonds with P-centered radicals, which can be easily generated through single-electron transfer (SET) or hydrogen atom transfer (HAT) using metal salts, peroxides, photocatalysts and others, has created a new avenue for the construction of organophosphorus compounds. As a consequence, the past few years have witnessed a renewed awareness regarding the utilization of P-centered radicals in constructing vitally important organophosphorus compounds[6-11]. The main characteristic of non-planar P-centered radicals with a variable degree of s-character (especially those containing P=O bonds) is their high reactivity in undergoing radical addition to unsaturated compounds [Figure 1][12]. In particular, the P-centered radical-based phosphorylation of organic compounds, mediated by transition metals or excessive oxidants, has witnessed significant advances. In addition, visible-light photoredox catalysis has emerged as a powerful means to achieve new reactions in organic chemistry under mild conditions [Figure 2]. Since
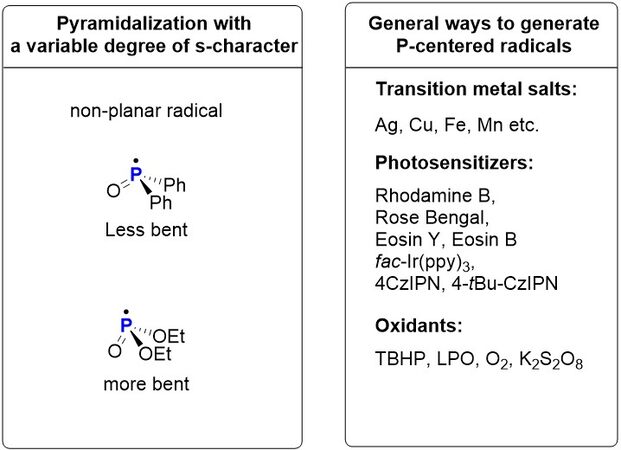
Figure 1. Geometries of P-centered radicals and general methods for generating P-centered radicals.
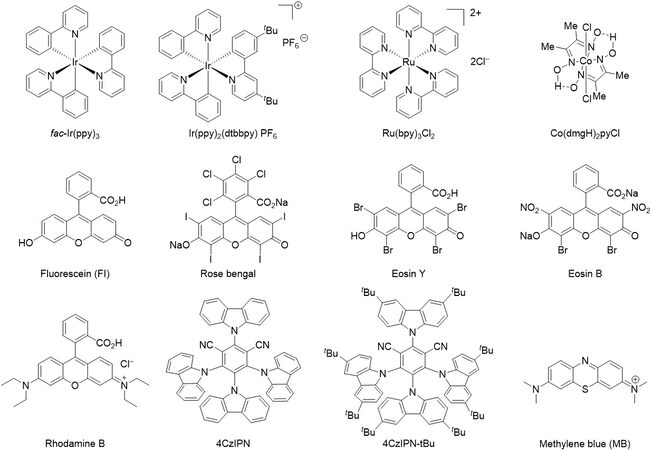
Figure 2. Structures of photocatalysts used in phosphorylation reactions.
Several reviews of phosphorylation have been published in recent years. For example, Gao et al.[6,8] reviewed the phosphorus radical-based difunctionalization of unsaturated compounds in 2017 and 2018. Cai et al.[9] published a review on visible light-mediated C-P bond formation reactions in 2019, while Chen et al.[10] summarized recent advances in the construction of phosphorus-substituted heterocycles in 2020. In 2021, Ung et al.[11] reviewed photomediated C-H phosphorylation reactions. Given that such reviews concentrate on relatively narrow research areas, there is currently no comprehensive review available on P-centered radical-based phosphorylation in organic chemistry integrating transition metal catalysis, photocatalysis and electrocatalysis. Moreover, many new works have been reported in the last five years. In this review, we summarize the recent advances in transition metal-catalyzed, photocatalytic and electrocatalytic C-P, N-P, S-P and O-P bond formation based on P-centered radicals. While this review mainly focuses on reactions reported in the past five years, some earlier pioneering works are also discussed.
PHOSPHORYLATION OF ALKENES
Alkenes, including styrenes, enamides and enolsilanes, are valuable and readily available feedstocks. The functionalization of alkenes, especially their difunctionalization, has developed into a highly versatile methodology for the building of highly functionalized skeletons in organic chemistry. Specifically, the P-radical-based functionalization of alkenes can be divided into the following categories [Figure 3]. The intermolecular phosphorylation process is initiated by radical addition to the carbon-carbon double bond with the generation of a carbon-centered radical intermediate, which can combine with an extrinsic radical donor, metal complexes or further be oxidized to a carbocation or reduced to a carboanion. In addition, the carbocation can also undergo β-H elimination to regenerate the alkenyl functionality. With regard to intramolecular functionalization, alkenes tethered with radical acceptors, nucleophilic or electrophilic groups dominate this reaction. Generally, after the addition of a P-centered radical to the alkenes, the subsequent carbon-centered radical, carbocation or carboanion may be trapped intramolecularly by a radical acceptor, nucleophilic or electrophilic groups, leading to the intramolecular cyclizing products.
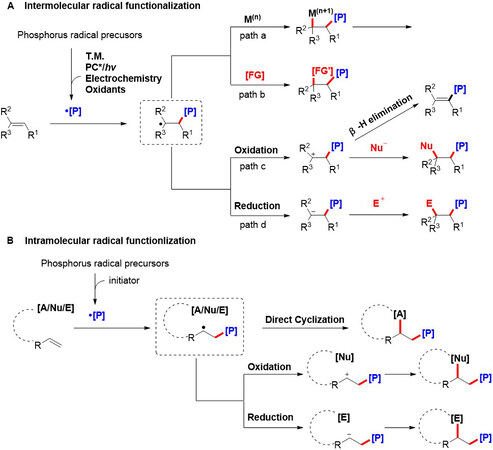
Figure 3. Radical functionalization of alkenes based on P-centered radicals.
Hydrophosphorylation of alkenes
The hydrofunctionalization of alkenes provides a facile route to a wide range of functionalized alkanes. In the last decade, the Lewis acid- or transition metal-catalyzed hydrophosphorylation of alkenes, especially of activated alkenes, has been significantly developed. However, the method for the hydrophosphorylation of unactivated alkenes through a radical pathway is still limited[16]. In 2013, Yoo et al.[17] described the visible-light photoredox-catalyzed hydrophosphinylation of unactivated alkenes with secondary phosphine oxides (SPOs) under mild reaction conditions using a commercially available and inexpensive organic dye as a photocatalyst and isopropanol as a green solvent [Figure 4]. This mild protocol is compatible with a variety of unactivated alkenes and functional groups, including alcohols, alkenyl ethers and so on. However, diaryl phosphine oxide containing a slightly more electron-rich substituent could only provide the corresponding product with a moderate yield. The mechanism is proposed as follows. The SET oxidation of phosphinous acid is promoted by the photoexcited Rhodamine B (PC*) to generate the corresponding radical cation. The deprotonation of such a radical cation generates a phosphinoyl radical, which undergoes radical addition to an alkene to form an alkyl radical intermediate. Regeneration of the phosphinoyl radical and the yielding of the desired hydrophosphinylation product could be realized by chain propagation from the HAT of SPO or its trivalent tautomer with the alkyl radical intermediate. Furthermore, the reduced photocatalyst PC•- may be quenched by minute amounts of oxygen gas in the solvent to regenerate the Rhodamine B (PC).
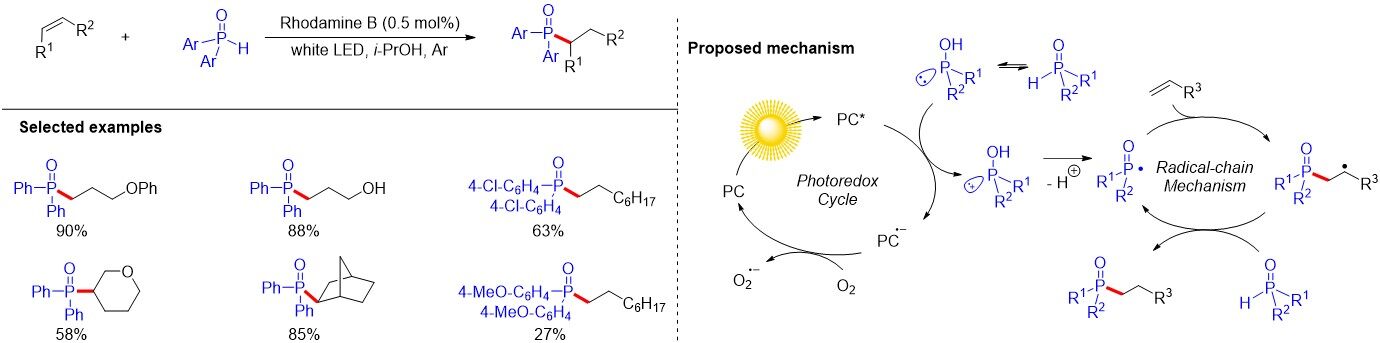
Figure 4. Hydrophosphinylation of unactivated alkenes with secondary phosphine oxides under visible-light photoredox catalysis.
Recently, based on previous works, Wang et al.[18] reported a P-centered radical-mediated anti-Markovnikov hydrophosphorylation of unactivated alkenes and remote alcohol oxidation through an intramolecular 1,5(6)-HAT procedure [Figure 5]. Notably, diverse sulfonyl- and malonate-substituted ketones or aldehydes could also be obtained from the alkenols. This mild and versatile reaction provides a general method to access diverse hydrofunctionalized products in moderate to good yields from a wide range of alkenols.
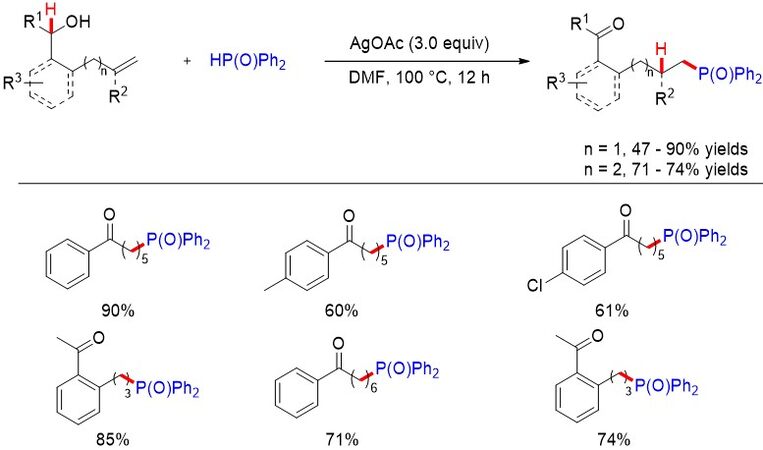
Figure 5. Anti-Markovnikov hydrofunctionalization of unactivated alkenes via a 1,5(6)-HAT procedure.
In addition, the visible light-driven hydrophosphinylation of unactivated alkenes was also explored by
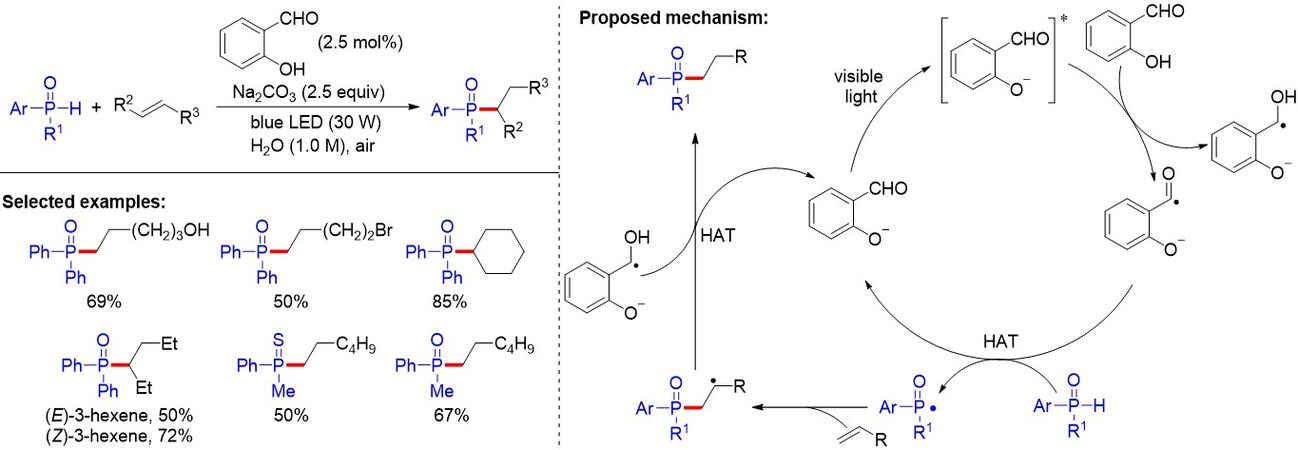
Figure 6. Visible light-induced salicylaldehyde-catalyzed hydrophosphinylation of unactivated alkenes.
Carbophosphorylation
Carbophosphorylation involves the construction of new C-P bonds with the simultaneous forging of C-C bonds, including C(sp3)-C(sp), C(sp3)-C(sp2) and C(sp3)-C(sp3) bonds. In this section, we divide carbophosphorylation into the two categories of intramolecular and intermolecular carbophosphorylation for simplicity.
Intramolecular carbophosphorylation
Aromatic organophosphorus compounds are ubiquitous and can be found in a wide range of pharmaceuticals, nucleotides and phosphine-containing ligands. Thus, the development of a concise and efficient method for C-P bond formation is hugely desirable. In 2012, Li et al.[20] reported a silver-catalyzed carbon-phosphorus difunctionalization of alkenes through a carbophosphorylation and C-H functionalization cascade process, leading to a diverse range of phosphorylated oxindoles through intramolecular direct annulation [Figure 7]. To understand the catalytic cycle of carbon phosphorylation reactions, high-resolution mass spectrometry was used to monitor the key intermediates. Mechanistic studies indicated that a silver-promoted generation of the phosphoryl radical might be involved in this radical cascade reaction. In 2014, a metal-free oxidative arylphosphination of activated N-substituted-N-arylacrylamide derivatives by a phosphorylation cascade was developed by Li et al.[21] to afford a variety of phosphorus-containing oxindole moieties.
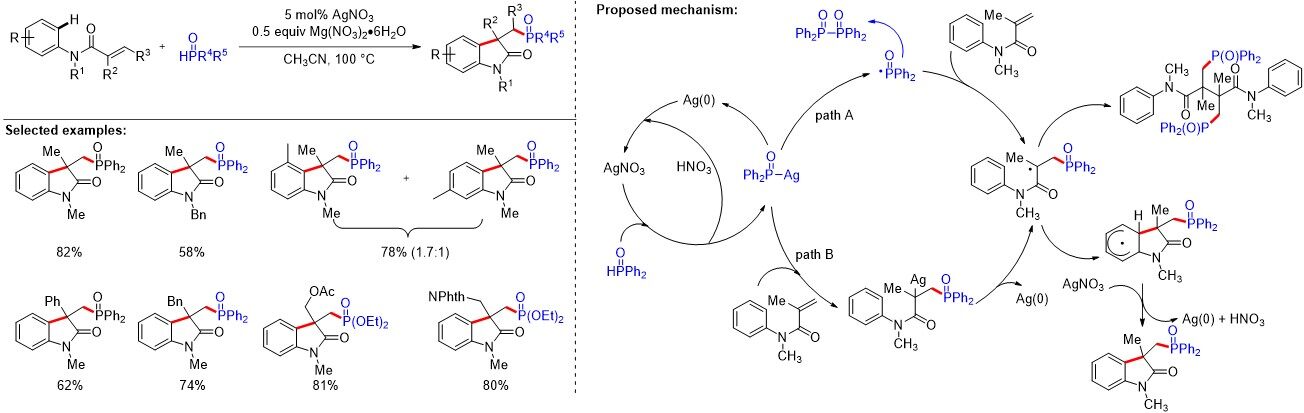
Figure 7. Silver-catalyzed carbon-phosphorus functionalization of alkenes.
The chroman-4-one framework is a structural motif that widely exists in numerous natural products and pharmaceuticals. Due to the importance of the chroman-4-one scaffold and organophosphorus compound, the synthesis of phosphorylated chroman-4-ones has attracted significant attention. Although considerable progress has been achieved in this area, the development of a transition metal-free approach for the synthesis of chroman-4-ones bearing phosphine oxide functionality remains highly desirable. In 2016,
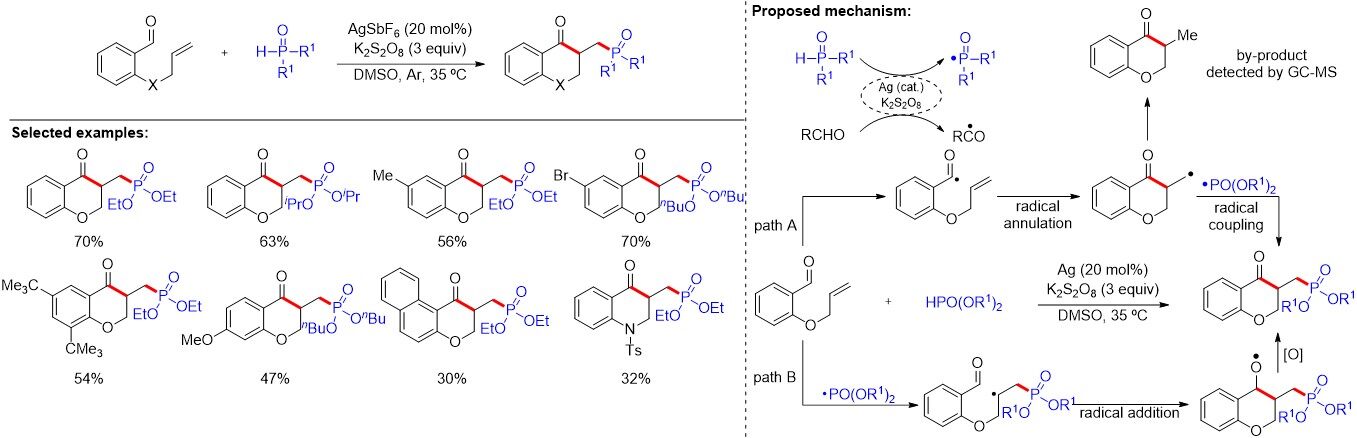
Figure 8. Radical cascade cyclization-coupling of 2-(allyoxy)arylaldehydes for the facile synthesis of functionalized chroman-4-ones.
Indoles, including oxindoles and indolines, represent a ubiquitous structural motif that constitutes a variety of natural alkaloids and clinically used drugs. The combination of the indole motif with a phosphorus group could result in a wide range of compounds exhibiting promising anti-tumor, anti-HIV and antimycobacterial bioactivities. Although a pioneering radical amino-phosphorylation of unactivated alkenes of 2-allylanilines toward 2-phosphonomethyl-N-sulfonyl indolines has been established by
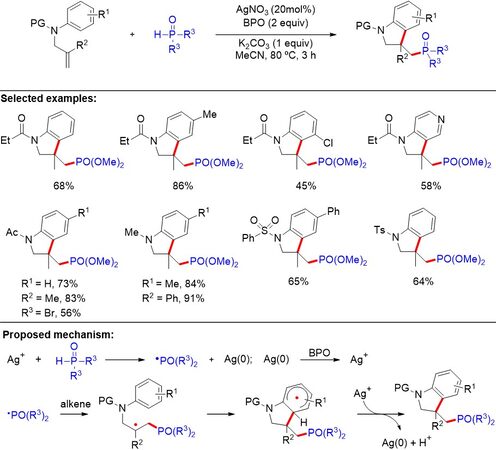
Figure 9. Silver-catalyzed radical arylphosphorylation of unactivated alkenes for the synthesis of 3-phosphonoalkyl indolines.
Dihydroisoquinolones have also been widely found in complex natural products, biomolecules and pharmaceuticals, and display extraordinary biological activities. By elegantly designing the substrates, in 2019, Liu et al.[26] reported a silver-mediated phosphorylation/cyclization of N-allylbenzamides with phosphine oxides for the synthesis of phosphoryl-substituted dihydroisoquinolones [Figure 10]. The method displays a broad substrate scope, good scalability and high efficiency under mild reaction conditions. As shown in Figure 10, apart from the application of stoichiometric silver salts as the oxidants, the reaction mechanism is similar to the above silver-catalyzed radical arylphosphorylation of unactivated alkenes.
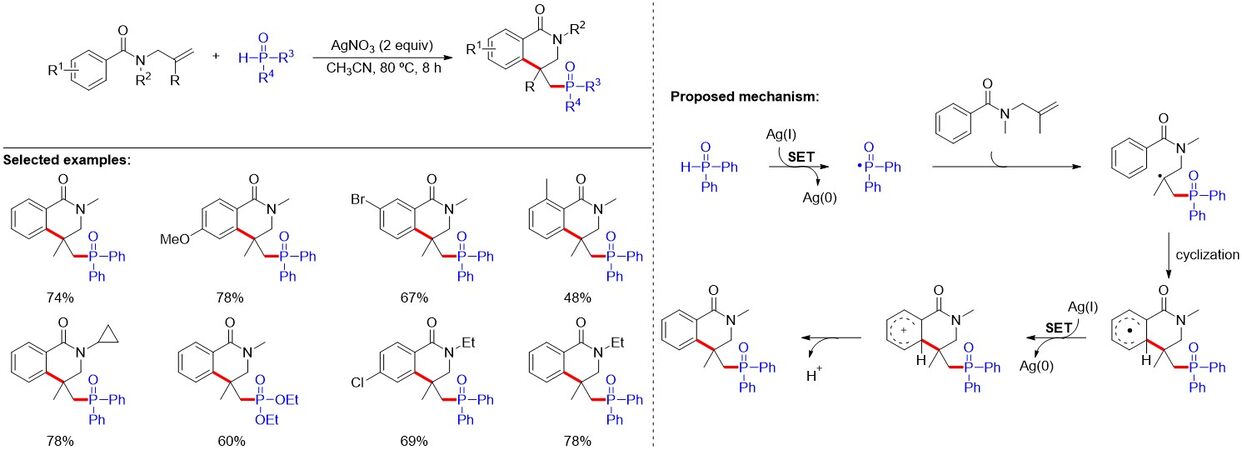
Figure 10. Silver-mediated radical phosphorylation/cyclization of N-allylbenzamides to access phosphoryl-substituted dihydroisoquinolones.
Despite the above achievements, transition metal reagents and/or high temperature are still required in these approaches. Thus, the development of more environmentally friendly and practical procedures to construct dihydroisoquinolone derivatives under room temperature is highly desirable. In 2019, Liu et al.[27] reported a visible light-promoted transition metal-free approach toward phosphoryl-substituted dihydroisoquinolones via a cascade phosphoarylation/cyclization of N-allybenzamides [Figure 11]. This approach features mild reaction conditions, convenient operation and a broad substrate scope. A series of Stern-Volmer fluorescence quenching experiments implied that the excited photocatalyst was effectively quenched by diphenylphosphine oxide. In addition, the quantum yield of the reaction was determined to be 0.26, precluding radical chain processes in this photochemical reaction.
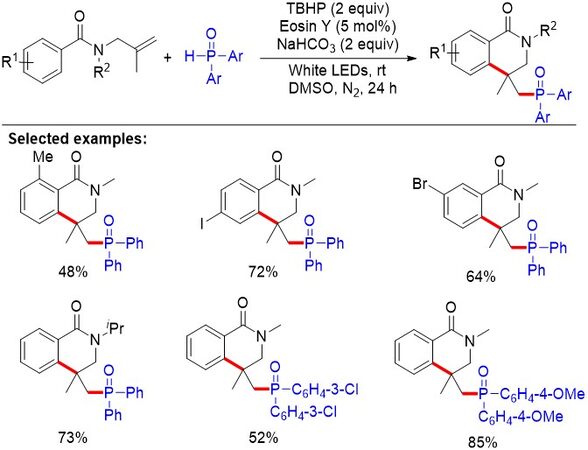
Figure 11. Visible light-promoted transition metal-free approach toward phosphoryl-substituted dihydroisoquinolones via cascade phosphorylation/cyclization of N-allylbenzamides.
Polyheterocycles are ubiquitous structural motifs widely found in natural products and pharmaceuticals that have attracted tremendous attention from chemists. In 2016, Liu et al.[28] reported a silver-catalyzed carbon-phosphorus functionalization for the synthesis of various phosphorylated polyheterocycles in moderate to good yields with high diastereoselectivity. Notably, the existence of P-centered radicals in the reaction was confirmed by electron paramagnetic resonance experiments. As depicted in Figure 12, the selective addition of the phosphorus radical to the α-position of the α, β-unsaturated carbonyl compound is crucial for the success of the reaction.
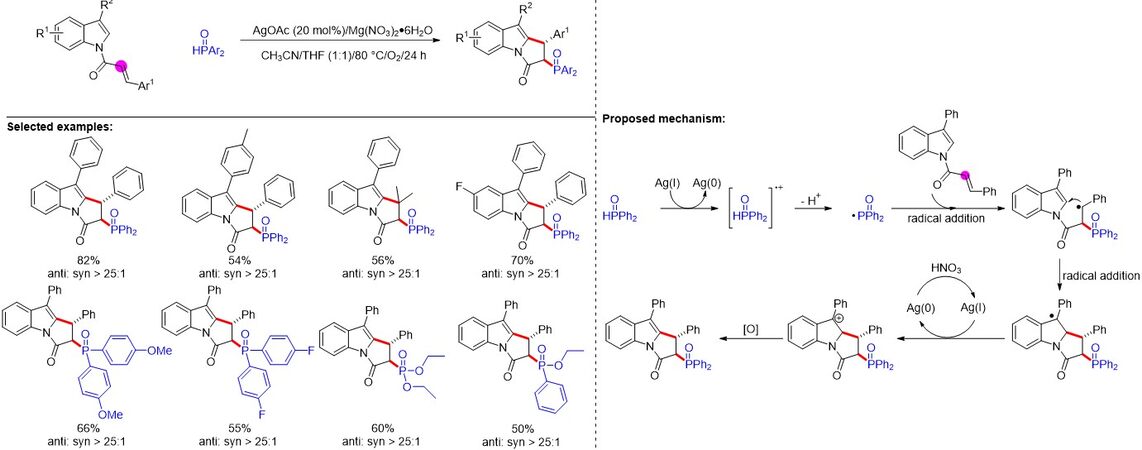
Figure 12. Silver-catalyzed carbon-phosphorus functionalization for polyheterocycle formation.
Later, a similar strategy was reported by Gorre et al.[29], who employed eosin Y as a photocatalyst to promote the metal-free phosphorus radical-mediated construction of phosphinoylpyrroloindoles [Figure 13]. This protocol features air as a green oxidant, high functional group tolerance, excellent chemo- and diastereoselectivity and mild reaction conditions. Importantly, the transformation proceeded through cascade C-P and C-C (5-endo-trig cyclization) bond formation with excellent diastereo- and chemoselectivity. The mechanism of the reaction is illustrated in Figure 13. Firstly, photoexcited eosin Y (EY*) oxidizes diphenylphosphine oxide via a SET to generate the radical cation. Upon deprotonation of the radical cation, the generated phosphinoyl radical is then added to the highly electron dense α-position of the carbonyl group, followed by 5-endo-trig cyclization, giving rise to the radical intermediate. Subsequently, the intermediate was oxidized to deliver the carbocation, which produces the final product upon protonation. Eventually, the photocatalytic cycle is closed after oxidation of the reduced photocatalyst (EY•-) by O2.
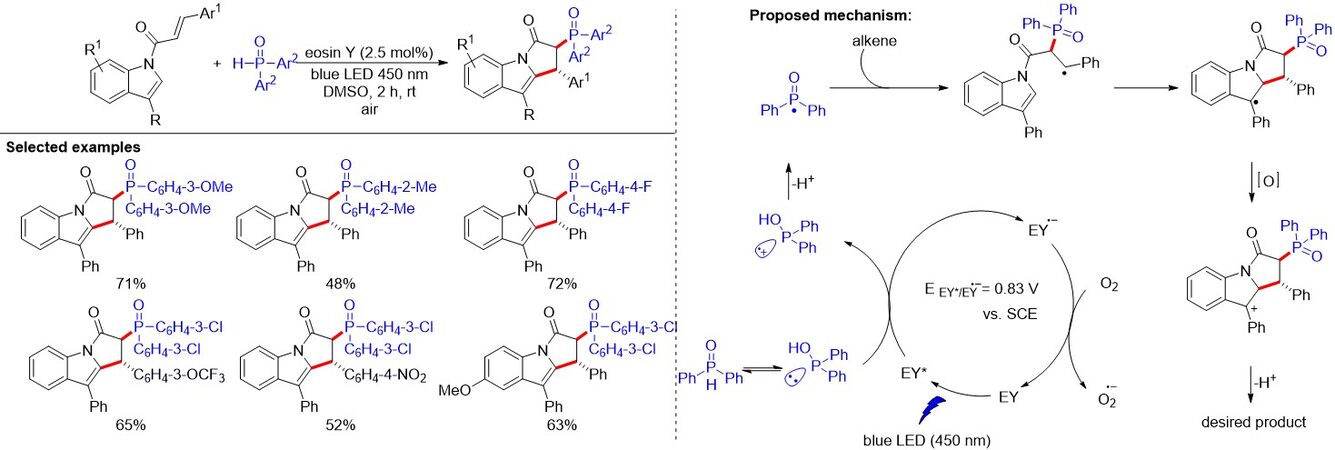
Figure 13. Visible light-driven metal-free aerobic synthesis of highly diastereoselective phosphinoylpyrroloindoles.
Phenanthridine derivatives represent an important category of scaffolds in biologically active molecules. The synthesis of functionalized phenanthridines starting from biphenylvinyl azides via a radical tandem cyclization strategy has achieved significant progress. Inspired by the works of alkylation and trifluoromethylation of vinyl azides, in 2017, Li et al.[30] described a visible light-induced tandem phosphorylation cyclization of vinyl azides for the synthesis of phosphorylated phenanthridines [Figure 14]. As illustrated in Figure 14, the addition of phosphoryl radicals to vinyl azide compounds in the reactions generate iminyl radicals, which undergo intramolecular homolytic aromatic substitution to deliver the final phenanthridines.
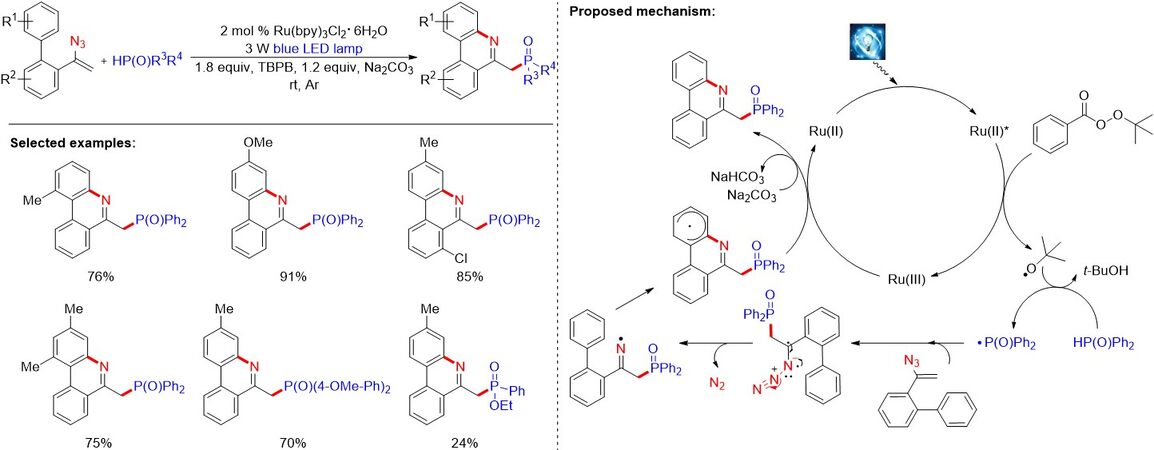
Figure 14. Visible light-induced tandem phosphorylation cyclization of vinyl azides.
Benzimidazo-isoquinoline-6(5H)-ones, as a class of important N-containing polycyclic scaffolds, have attracted notable research interest. In 2019, Sun et al.[31] described an efficient protocol for the synthesis of various phosphoryl-substituted benzimidazo[2,1-a]isoquinoline-6(5H)-ones from 2-arylbenzoimidazoles and phosphine oxides, including various diphenylphosphine oxides, ethyl phenylphosphinate and dialkyl H-phosphonates, using a AgNO3/K2S2O8 catalytic system. The reaction features a wide substrate scope, mild reaction conditions and easy scale up, thereby providing a straightforward method for the synthesis of valuable phosphoryl-substituted benzimidazo[2,1-a]isoquinoline-6(5H)-ones [Figure 15]. A Mn(III)-promoted tandem phosphinoyl/cyclization of 2-arylindoles/2-arylbenzimidazoles with disubstituted phosphine oxides was also developed by Jiang et al.[32]. As illustrated in Figure 15, this reaction mechanism is similar to the above-discussed phosphorus radical-initiated radical cascade reactions.
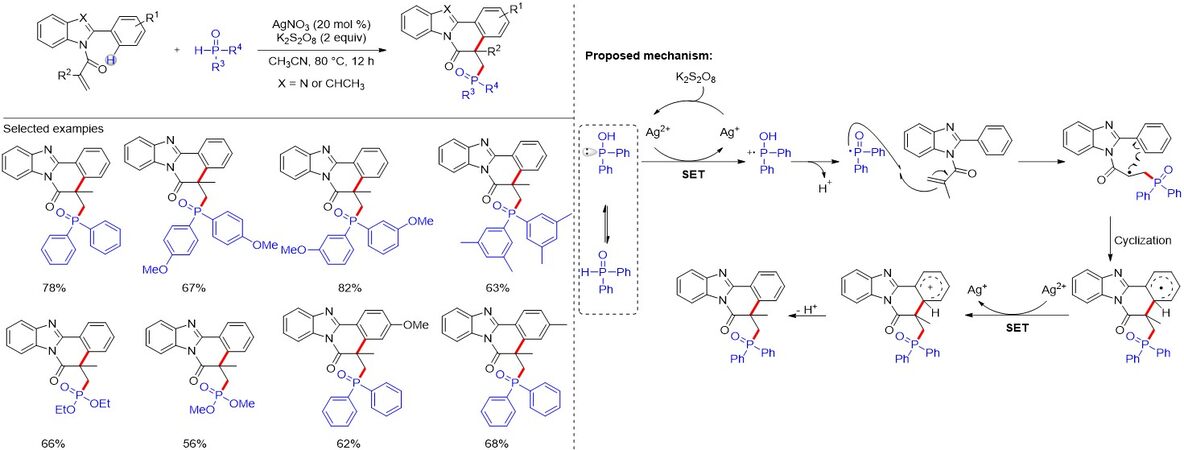
Figure 15. Silver-catalyzed synthesis of phosphoryl-substituted benzimidazo[2,1-a]isoquinoline-6(5H)-ones.
Most radical cyclization reactions involving phosphorus radicals are based on an oxidative process. In contrast, the redox-neutral phosphorylation/cyclization pathway remains less explored. Based on our previous work[33] of the visible light-induced phosphonocarboxylation of alkenes, in 2021, we disclosed the visible light-driven phosphonoalkylation of alkenes tethered with alkyl sulfonates to construct phosphorylated three-membered carbocyclic scaffolds with H-P(O) compounds [Figure 16][34]. This transition metal-free protocol exhibits good functional group tolerance, a broad substrate scope and high yields under mild reaction conditions. As shown in Figure 16, the intramolecular nucleophilic substitution reaction is the key step to furnishing the final cyclized product.
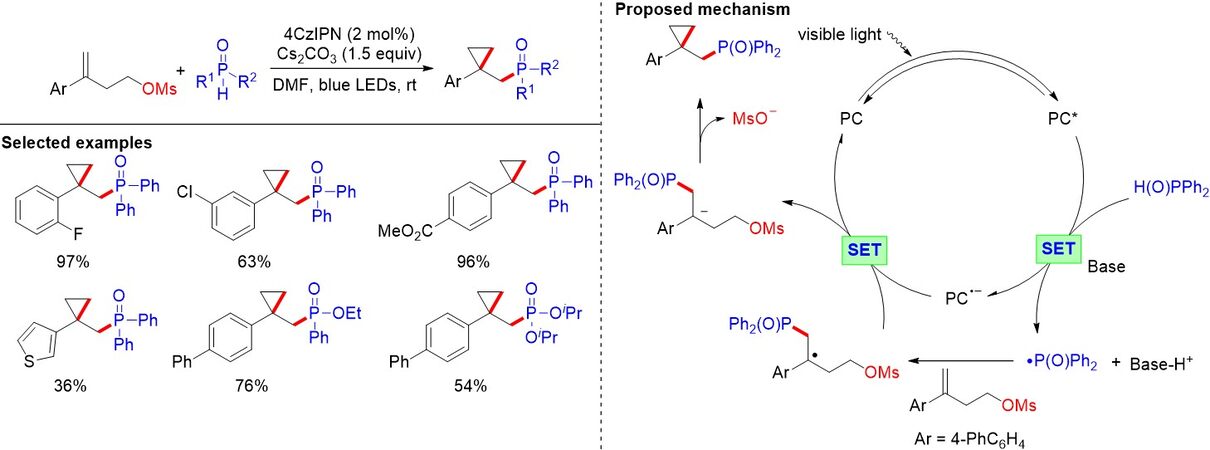
Figure 16. Visible-light-driven phosphonalkylation of alkenes.
Phosphinylation with concomitant 1,n-migration of a functional group represents a powerful strategy for building structurally difficult to access scaffolds. In 2014, Chu et al.[35] reported a silver-catalyzed radical phosphinylation of α, α-diaryl allylic alcohols with diarylphosphine oxides for the convenient synthesis of α-aryl-β-phosphinylated carbonyl ketones in moderate to good yields through chemoselective 1,2-aryl migration, involving the formation of C(Ar)-C(sp3) and C(sp3)-P bonds in a single step [Figure 17]. In the same year, similar work with H-phosphonates as the radical precursors was proposed by Mi et al.[36].
Figure 17. Silver-catalyzed carbophosphonation of α, α-diaryl allylic alcohols for the synthesis of β-aryl-γ-ketophosphonates.
Although approaches for the construction of β-aryl-γ-ketophosphine oxides via a radical carbophosphinylation of allylic alcohols have been described, the methods available still suffer from the need for expensive transition metal salts and high reaction temperatures. Inspired by these precedents,
Figure 18. Eosin Y-catalyzed visible light-promoted carbophosphinylation of allylic alcohols via a radical neophyl rearrangement.
As the generation of phosphorus radicals is frequently accompanied by stoichiometric amounts of radical initiators or external oxidants, the design and synthesis of new phosphorus radical precursors remain important areas of research interest. In 2018, Li et al.[38] reported a novel type of phosphine radical precursor, namely, an oxime phosphonate, which was synthesized and applied in the visible light-catalyzed phosphonation-annulation of unsaturated sulfonamides and carboxylic acids, affording a variety of β-phosphonopyrrolidines and β-phosphonolactones. Notably, additional oxidants and bases were not needed in the reaction. Recently, this novel radical precursor was applied in a semi-pinacol rearrangement reaction for the synthesis of γ-oxo-phosphonates by Wang et al.[39] [Figure 19]. Importantly, the reaction avoids using external oxidants. As displayed in Figure 19, the proposed mechanism for the semi-pinacol rearrangement reaction commences with a photoinduced SET between oxime phosphonate and excited-state *Ir(III), leading to the formation of the phosphorus radical. The addition of the P-centered radical to the alkene of the substrate then rapidly gives the alkyl radical, which immediately generates a benzyl radical via the semi-pinacol rearrangement process. Finally, the resulting benzyl radical was oxidized by the Ir(IV) complex to form a benzyl cation, which is converted to the ultimate product after deprotonation.
Figure 19. Photoinduced phosphorus radical-based semi-pinacol rearrangement reaction for the synthesis of γ-oxo-phosphonates.
Although the difunctionalization of unactivated alkenes with radical 1,2-aryl migration provides an efficient method for accessing highly functionalized products, the radical 1,2-alkynyl migration has rarely been studied. Recently, on the basis of a sequential oxidation mechanism, Jin et al.[40] reported a silver-promoted phosphonation/alkynylation of alkenes proceeding with radical 1,2-alkynyl migration, affording a range of α-alkynyl γ-ketophosphine oxides in moderate to good yields [Figure 20]. It is noteworthy that the addition of the P-centered radical to the alkenes initiated 1,2-alkynyl migration via 3-exo-dig cyclization.
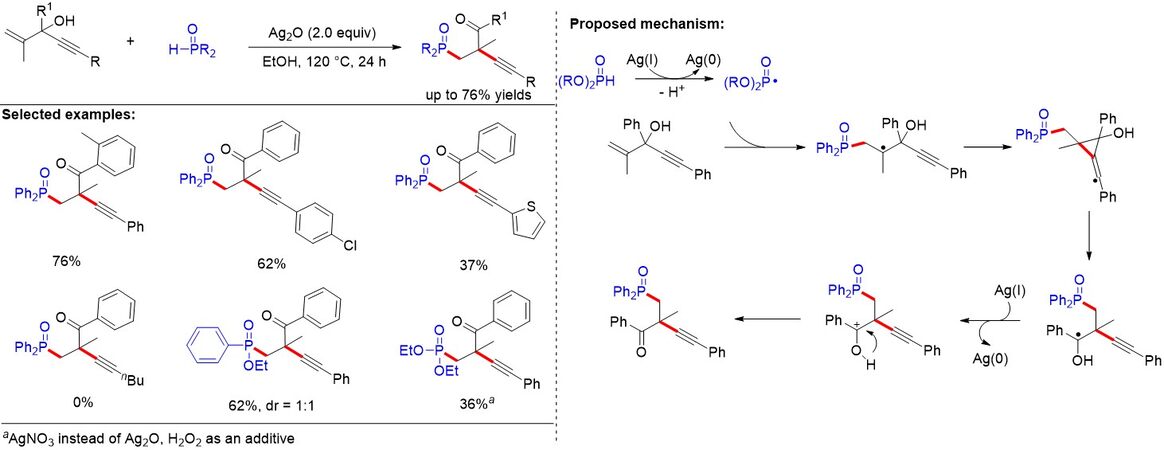
Figure 20. Silver-promoted phosphonation/alkynlation of alkenes proceeding with radical 1,2-alkynyl migration.
Intermolecular carbophosphorylation
Although intermolecular reactions are more challenging than intramolecular reactions, they remain efficient pathways for the difunctionalization of alkenes. Cyano is a versatile group that can be converted to a diverse array of functionalities, such as amides, acids, aldehydes and heterocycles, for the building of structurally intriguing bioactive compounds. In 2017, Zhang et al.[41] reported a cyanophosphinoyl of alkenes using Mn(OAc)3 as an oxidant to generate the P-centered radical and CuCN as a catalyst to facilitate the cyanation process [Figure 21]. The reaction is performed under mild conditions, tolerating both aromatic and aliphatic alkenes to afford vicinal cyanophosphinoylation products with high regioselectivity. The cyano group in the product can be easily transformed to other valuable compounds of synthetic and biological interest.
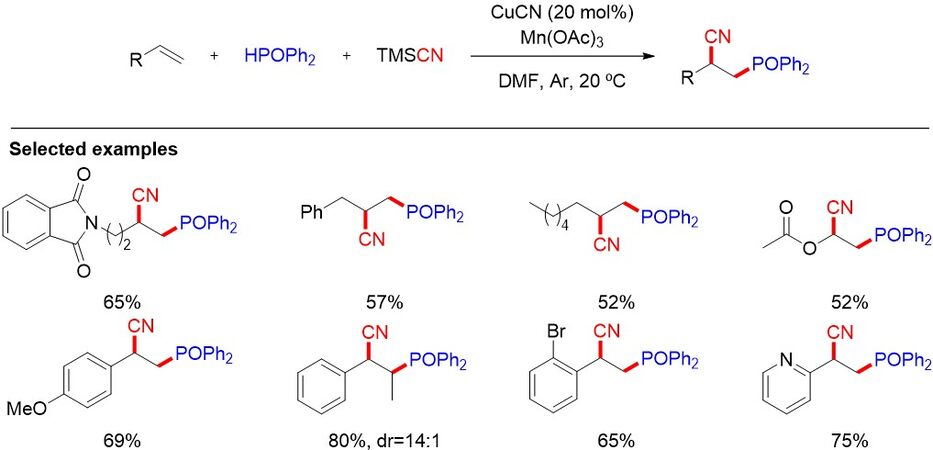
Figure 21. Cyanophosphinoyl of alkenes.
Although a series of phosphonylation-based difunctionalization reactions of alkenes have been developed, phosphorus radical-initiated asymmetric reactions have seldom been reported owing to the involvement of highly reactive carbon-centered radical intermediates. In 2019, Zhang et al.[42] collaboratively reported a copper-catalyzed enantioselective phosphinoylcyanation of styrenes via a tandem radical relay, allowing for the direct and efficient synthesis of various β-cyanodiaryphosphine oxides [Figure 22]. Preliminary mechanistic studies demonstrate that t-BuOOSiMe3, generated in situ from t-BuOOH, serves as a radical initiator to trigger t-butoxy radical production upon oxidization of the (L5)CuICN species via a proton-coupled electron transfer (PCET) pathway, leading to the generation of phosphinoyl and benzyl radicals in sequence. A series of chiral γ-aminophosphine ligands can be easily obtained from the β-cyanodiarylphosphine oxides.
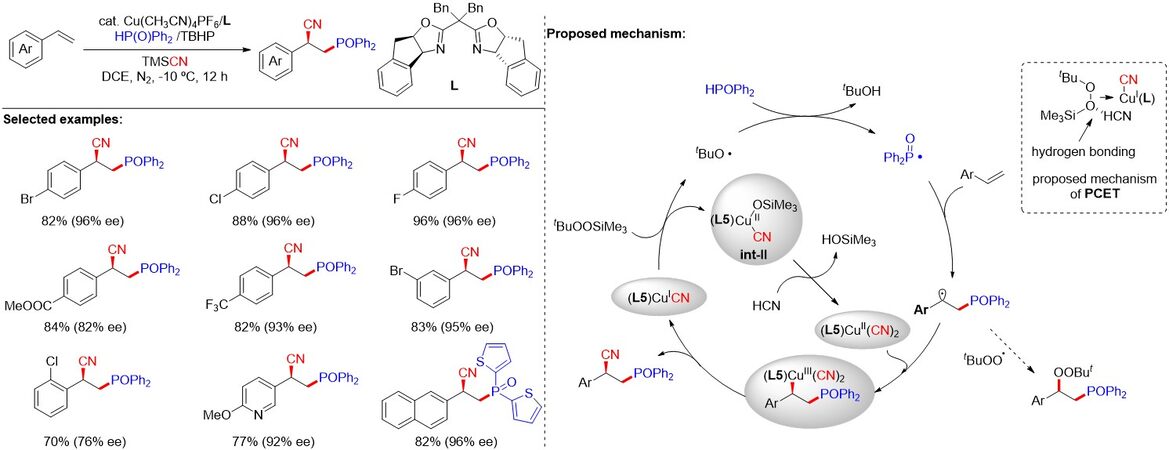
Figure 22. Proton-coupled electron transfer enables tandem radical relay for asymmetric copper-catalyzed phosphinoylcyanation of styrenes.
β-Pyridylphosphine motifs consisting of a pyridine with a hard σ-donor feature and a phosphine with a soft π-acceptor are widely employed as versatile ligands for designing transition metal catalysts. However, the simultaneous incorporation of both phosphine and pyridyl moieties into unsaturated C-C bonds remains an unresolved challenge. In 2018, He et al.[43] developed a one-pot difunctionalization of unactivated alkenes with P(O)-H compounds and N-methoxypyridinium salts for the preparation of a variety of synthetically and biologically important β-pyridyl alkylphosphonates [Figure 23]. The authors proposed that the addition of a phosphonyl radical to the alkene to form the alkyl radical intermediate, which adds to the N-methoxypyridinium salt, is involved in the reaction. Density functional theory calculations were implemented to illustrate the solvation effect and the detailed mechanism of the reaction. Notably, other pathways leading to the byproducts are discussed for the reaction.
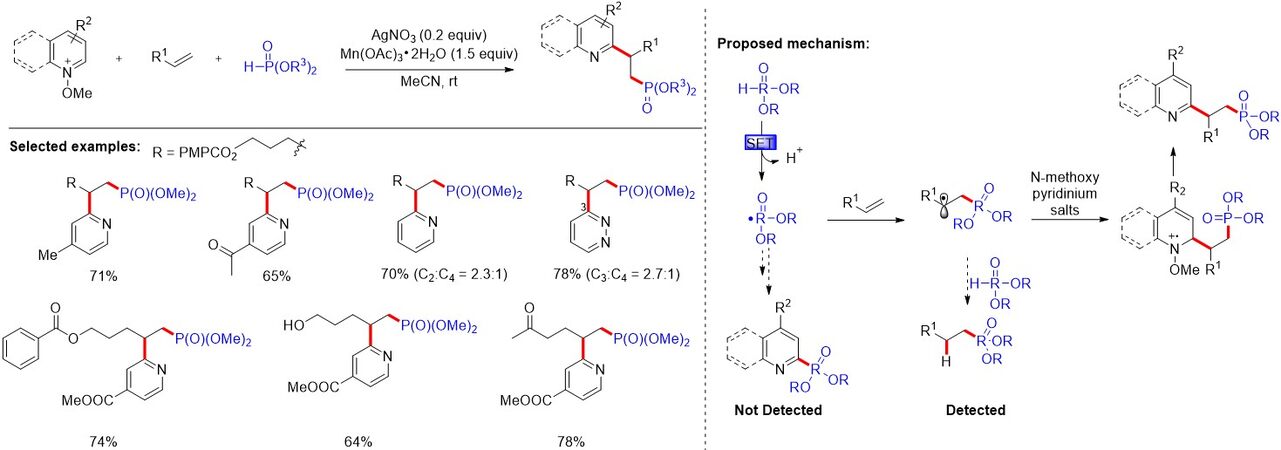
Figure 23. Difunctionalization of unactivated alkenes for the synthesis of β-pyridyl alkylphosphonates.
In contrast to the above work using silver salts and stoichiometric metal oxidants, a photocatalytic polarity-reversing radical cascade strategy for the difunctionalization of alkenes was proposed by Buquoi et al.[44] [Figure 24]. The phosphorus group and a heteroarene, such as pyridine, are concurrently added onto an olefin, which is enabled by the visible light-induced generation of electrophilic P-centered radicals. The methods presented serve as an inspiration for exploiting more multicomponent couplings based on radical polarity reversal. Notably, Lear et al.[45] also expanded polarity-reversed radical cascades to a variety of radical precursors, including N3, P(O)R2 and CF3, by employing hypervalent iodine reagents.
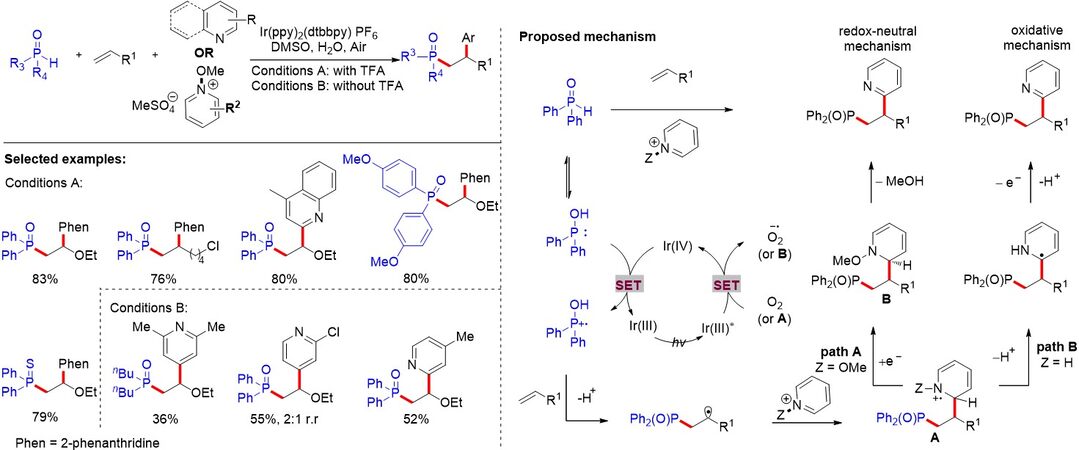
Figure 24. Heteroarene phosphinylalkylation via a catalytic polarity-reversing radical cascade.
While pyridylation/phosphinoylation as a new route for the 1,2-difunctionalization of alkenes at the C-2 position of pyridines has been realized, the alkylation of pyridines at the C-4 position remains unresolved. In 2021, Shen et al.[46] described the intermolecular pyridylation/phosphinoylation of alkenes at the C-4 position of pyridines using 4-cyanopyridines and diphenylphosphine oxides under photoredox conditions [Figure 25]. Mechanistic studies indicate that triethylamine serves as both a HAT and SET reagent via visible-light photoredox catalysis. Condition screening shows that a photocatalyst is essential for this reaction. Aromatic and aliphatic olefins could be converted to the corresponding products with moderate to good yields. A possible mechanism is proposed in Figure 25. Et3N (Eox1/2 = +0.83 V vs. Saturated Calomel Electrode (SCE) can be oxidized by PC* (Eox1/2 PC*/PC•- = +1.35 V vs. SCE) generated from the ground-state photocatalyst under visible-light irradiation. A SET between Et3N and excited-state PC* could then produce Et3N•+ and PC•-. The HAT between diarylphosphine oxide and Et3N•+ provides Et3NH+ and a phosphinoyl radical. Subsequently, the phosphinoyl radical undergoes facile radical addition to the alkenes, leading to a carbon radical. Simultaneously, the reduced PC•- (Ered1/2 PC/PC•- = -0.75 V vs. SCE in TFE) reduces 4-cyanopyridine (Ered1/2 = -0.62 V vs. SCE in TFE) via a SET to afford a persistent radical anion along with the regeneration of PC. Finally, the fusion of the C-C bond via intermolecular radical-radical coupling gives rise to the anion intermediate, which undergoes rapid rearomatization to afford the desired product along with the elimination of a cyanide anion.
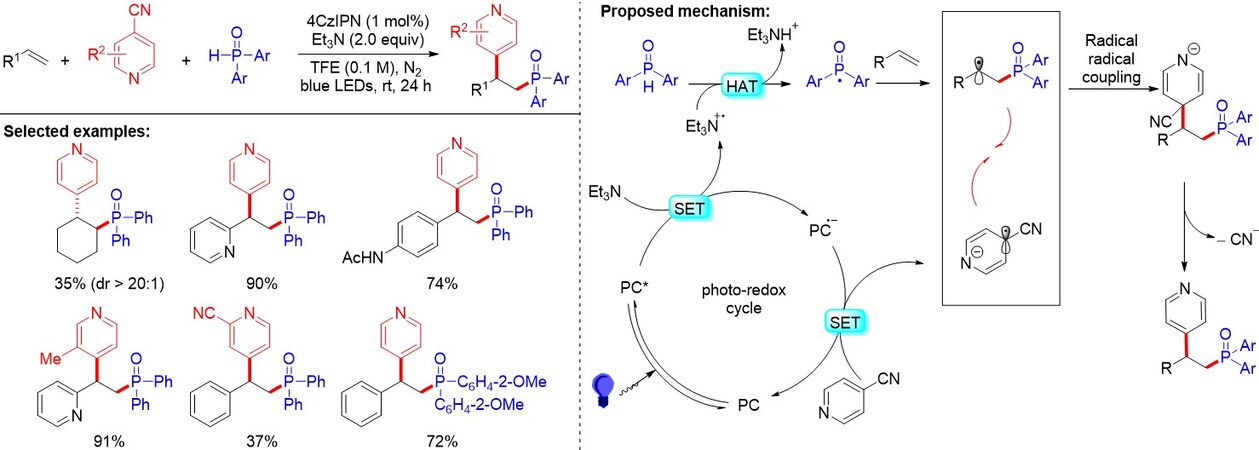
Figure 25. Metal-free visible light-induced photoredox catalyzed intermolecular pyridylation/phosphinoylation of alkenes.
Both phosphonyl and carboxyl are valuable groups and their simultaneous incorporation into organic molecules can serve as an efficient method to access pharmaceutically important β-phosphono carboxylic acids. In 2020, Fu et al.[33] provided a method for the phosphonocarboxylation of alkenes with carbon dioxide via visible-light photoredox catalysis [Figure 26]. This protocol features low catalyst loading, high regio- and chemoselectivities and good functional group tolerance. Furthermore, this sustainable, general and practical strategy generates important β-phosphono carboxylic acids, containing structurally complex unnatural α-amino acids. A wide range of alkenes, including enamides, styrenes, enolsilanes and acrylates, could be transformed to the desired products efficiently under mild reaction conditions. As presented in Figure 26, the SET between H-P(O) compounds and a photoexcited photocatalyst with the assistance of a base generates the phosphonyl radicals, which were captured by the C=C double bonds of enamides to selectively form the α-amido radical stabilized by phenyl and amide groups. A subsequent SET reduction by the reduced photocatalyst then affords the α-amido carbanions, which can react with CO2 to deliver the desired β-phosphono α-amino acids. Notably, the reaction outcompetes several side reactions, such as C-H bond carboxylation, hydrocarboxylation and hydrophosphinylation, as well as nucleophilic addition reactions between enamides, which could tautomerize to imines and H-P(O) compounds.
Figure 26. Transition metal-free phosphonocarboxylation of alkenes with carbon dioxide via visible-light photoredox catalysis.
Oxyphosphorylation of alkenes
β-Ketophosphine oxides are valuable phosphorous-containing compounds that exhibit a diverse range of applications in various synthetically useful transformations, especially as synthetic precursors to access olefins through the well-known Horner-Wittig reaction[47]. In 2011, the first catalytic and direct oxyphosphorylation of alkenes leading to β-ketophosphonates was reported by Wei et al.[48] using Cu(II) and Fe(III) as catalysts and avoiding the use of stoichiometric organometallic reagents [Figure 27]. A preliminary mechanistic investigation suggested that the carbonyl oxygen originates from dioxygen and this method involves a radical process. Since then, a variety of methods have been explored for the oxyphosphorylation of alkenes. For example, Zhou et al.[49] disclosed a domino Knoevenagel-decarboxylation-alkene difunctionalization sequence for the construction of β-ketophosphonates in moderate to good yields via the oxyphosphorylation of in situ generated cinnamic acids from easily available benzaldehydes and H-phosphonates by a cooperative Cu and Fe catalytic system in 2015 [Figure 28].
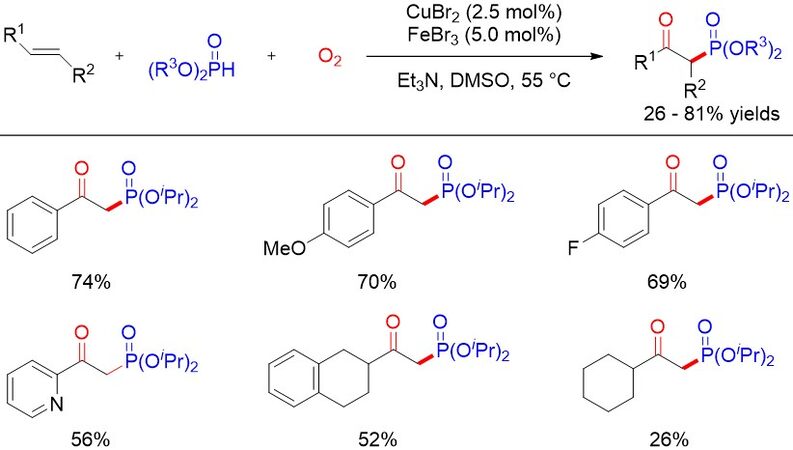
Figure 27. Cu- and Fe-cocatalyzed oxyphosphorylation of alkenes.
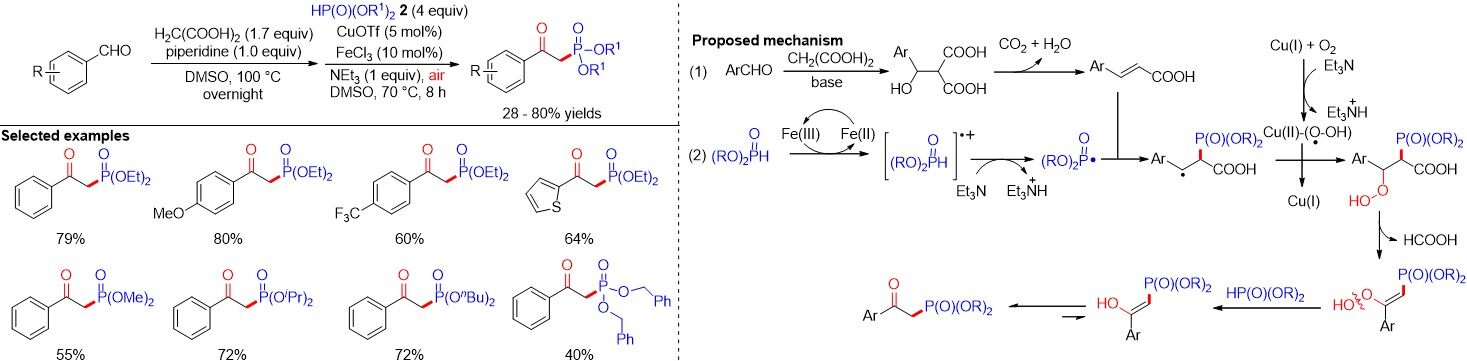
Figure 28. Cu- and Fe-cocatalyzed formation of β-ketophosphonates by a domino Knoevenagel-decarboxylation-oxyphosphorylation sequence.
In 2017, Chen et al.[50] described an efficient one-pot synthesis of β-ketophosphonates from cinnamic acids or alkenes with H-phosphonates and dioxygen via Cu-based catalysis [Figure 29]. This reaction is suitable for alkenyl carboxylic acids and alkenes, as well as a series of organophosphorus reagents, including diphenylphosphine oxide, dialkyl H-phosphonates and ethyl phenylphosphinate. The authors proposed that an active oxygen complex [(MeCN)nCuII-O-O] was involved in the reaction.
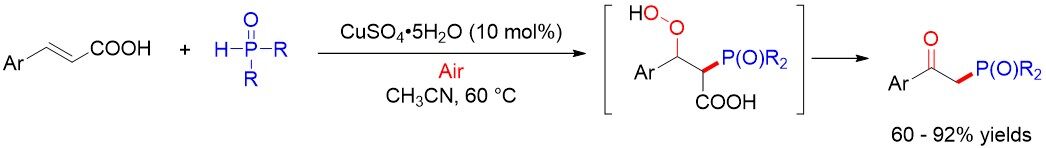
Figure 29. Oxyphosphorylation of alkenyl acids or alkenes.
Owing to their continuing interest in the efficient synthesis of β-ketophosphine oxides, Fu et al.[51] developed efficient methods for the synthesis of β-ketophosphine oxides from ketones in 2017 [Figure 30]. Such a copper-catalyzed aerobic oxidative coupling of ketones with P(O)-H compounds leads to a series of β-ketophosphine oxides with good functional group tolerance assisted by TBSOTf (tbutyldimethylsilyl triflate). Isotope-labelling experiments indicate that the carbonyl oxygen atom of the products originates from dioxygen. Firstly, the reaction is initiated by a SET between Ph2P(O)H and Cu(II) species to form diphenyl phosphine oxide cation radical and Cu(I) species. With the help of Et3N, the deprotonation generated the P-radical, which attacked enolsilane to give the β-phosphoryl alkyl radical. Afterwards, the unstable radical reacted with superoxide radical O2•- and Et3NH+ to deliver a hydroperoxide intermediate. Subsequently, the intramolecular elimination of R3SiOH in the presence of Et3N gave a hydroperoxide species, which was further reduced by diphenylphosphine oxide to give the enolate, which was eventually transformed into the product via a rapid tautomerization.
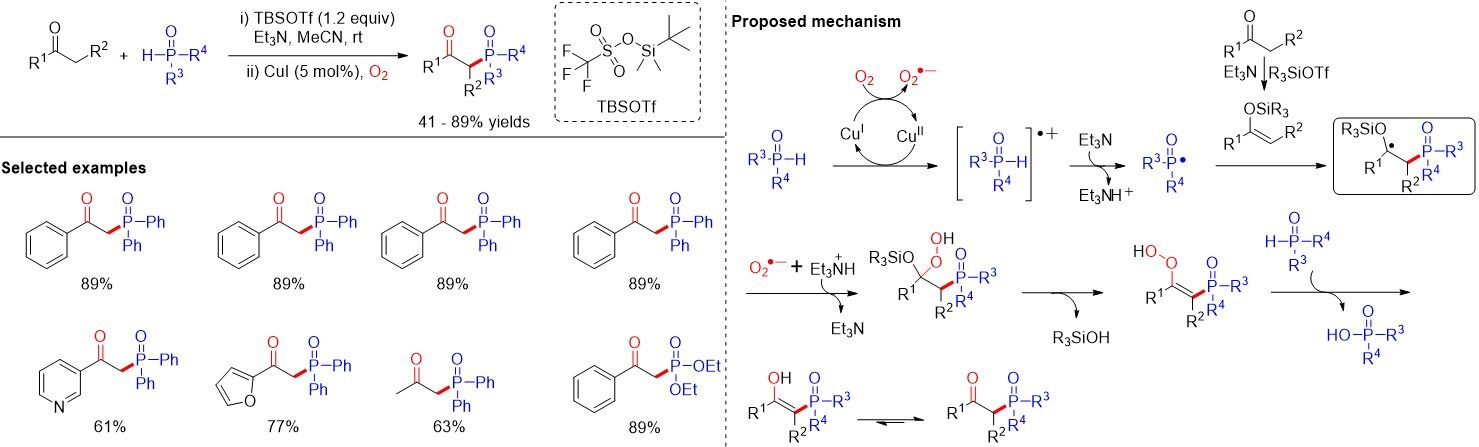
Figure 30. Copper-catalyzed phosphorylation of ketones assisted by silylating reagent.
In the same year, Zhang et al.[52] reported the copper-catalyzed one-pot synthesis of β-ketophosphine oxides from ketones and H-phosphine oxides, in which vinylhydrazinedicarboxylate was the key intermediate [Figure 31A]. Soon after, Liang et al.[53] demonstrated a copper-catalyzed oxyphosphorylation of enamides with P(O)-H compounds and dioxygen, allowing for the synthesis of a series of valuable β-ketophosphine oxides/β-ketophosphonates in moderate to good yields [Figure 31B]. Later, Nan and Yue[54] presented an additive-free method using CuCN as a catalyst and dioxygen as an external oxidant to provide a convenient and cost-effective approach for diverse β-ketophosphine oxides from alkenes in moderate to good yields (47%-72%) [Figure 31C]. In addition, Wang et al.[55] reported the hydrochloric acid-promoted Cu- and Fe-cocatalyzed deesterificative oxyphosphorylation of 2-substituted acrylates with H-phosphine oxides. Notably, similar reaction mechanisms were proposed for the above works. In contrast to previous studies, in 2019, Feng et al.[56] described a copper-catalyzed oxidative radical coupling/fragmentation reaction, giving rise to diverse β-ketophosphine oxides from simple allylic alcohols and H-phosphine oxides [Figure 31D]. Remarkably, an unprecedented three-component oxidative radical coupling/fragmentation processes on the basis of a series of control experiments was proposed.
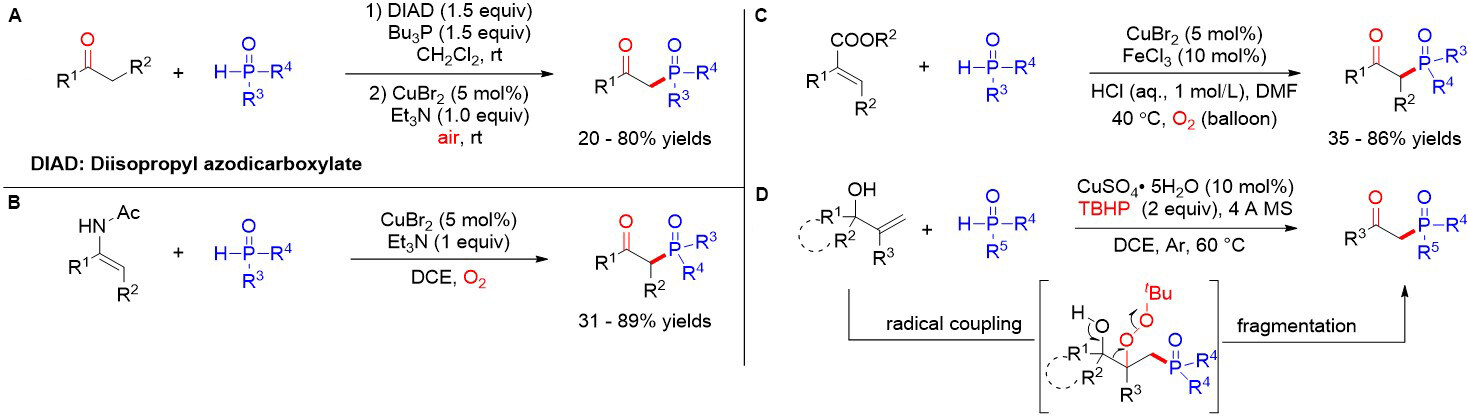
Figure 31. Oxyphosphorylation of enolsilane, enamide, cinnamic acid and allylic alcohol derivatives.
Concurrently, Gu et al.[57] reported a temperature-controlled chemoselective dehydrogenative phosphorylation or oxyphosphorylation of alkenes with H-phosphine oxides via copper-/iron-based catalysis. In addition to vinylphosphonates, β-ketophosphonates could also be obtained in moderate to good yields depending on the reaction temperature [Figure 32]. Mechanistic studies indicate that when the reaction temperature is 90 °C, the formed β-phosphoryl alkyl radical interacts with the hydroxyl radical, which is generated from Cu(I) with DTBP and H2O, to form β-hydroxyl phosphonate, which undergoes oxidation to give the desired β-ketophosphonates. When the reaction temperature is 110 °C, the formed β-phosphoryl alkyl radical is believed to undergo direct oxidation and following deprotonation delivers the vinylphosphonate product.
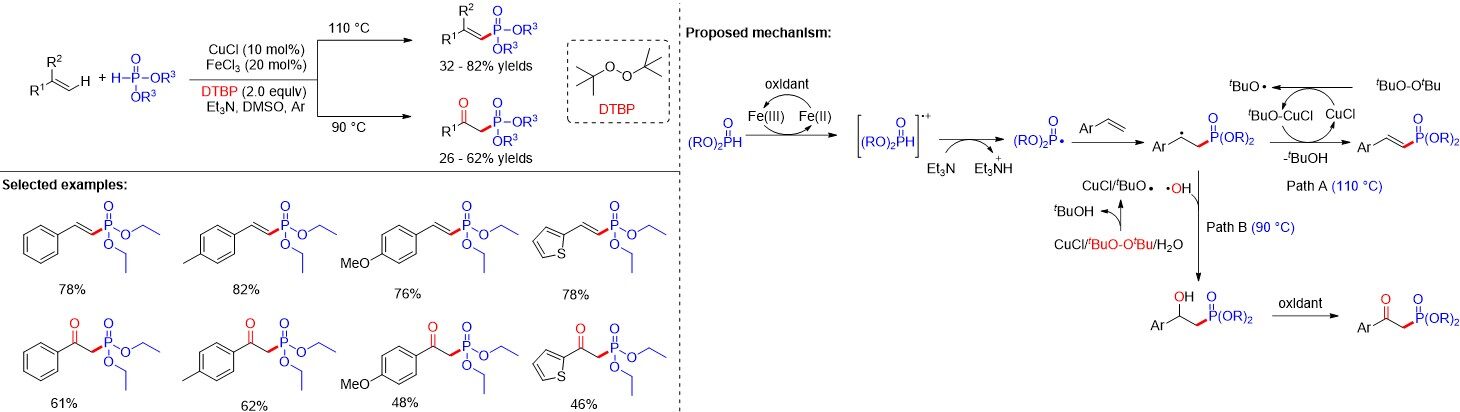
Figure 32. Copper/iron-catalyzed C-P cross-coupling of styrenes with H-phosphine oxides.
As well as homogeneous catalysis, heterogeneous catalysis also contributes greatly to the oxyphosphorylation of alkenes. In 2020, Moghaddam et al.[58] disclosed a typical copper ferrite nanoparticle (CuFe2O4)-catalyzed formation β-ketophosphonates in good to excellent yields via the oxyphosphorylation of styrenes with H-phosphonates [Figure 33]. Notably, the catalyst could be reused six times without a significant decrease in the yields and FESEM images revealed that the morphology of the catalyst remained constant. The reaction mechanism proposed by the authors is similar to the above-mentioned oxyphosphorylation of alkenes.
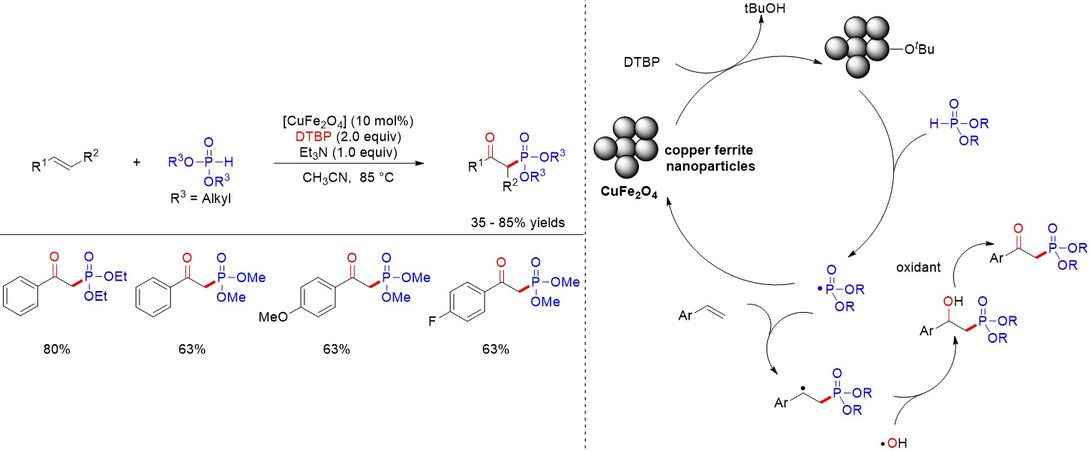
Figure 33. Copper ferrite nanoparticle-catalyzed formation of β-ketophosphonates.
Despite excellent progress in the synthesis of β-ketophosphine oxides, the development of more environmentally friendly, green and efficient methods for their synthesis remains highly desirable. In 2018, Shi et al.[59] developed the visible light-induced oxyphosphorylation of simple alkenes with H-phosphine oxides under ambient air [Figure 34]. In this reaction, a low-cost organic dye was used as the photocatalyst, providing an array of β-ketophosphine oxides with good yields under mild conditions.
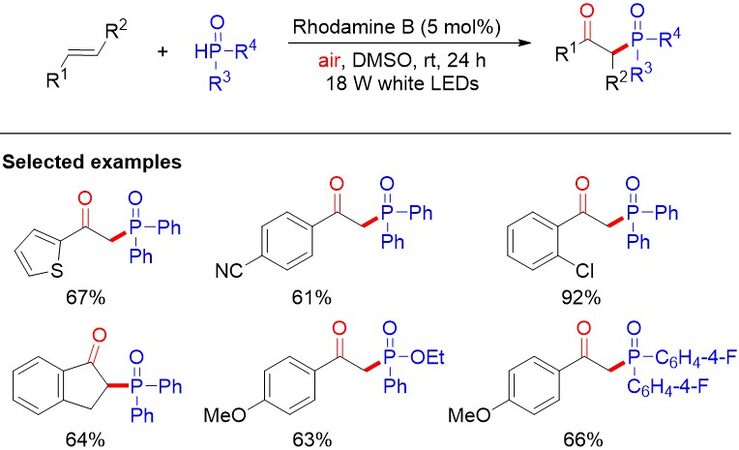
Figure 34. Visible light-promoted metal-free aerobic oxyphosphorylation of olefins as a facile approach to β-ketophosphine oxides.
As a result of their stability and accessibility, cinnamic acids have been employed as viable precursors to construct a various of valuable compounds through the extrusion of carbon dioxide. Qian et al.[60] reported the visible-light photoredox-catalyzed decarboxylative oxyphosphorylation of cinnamic acids with diarylphosphine oxides to afford the corresponding β-ketophosphine oxides under mild conditions [Figure 35]. Nevertheless, α- or β-methyl-substituted alkenes could not be tolerated in such a reaction. Initially, the oxidation of the excited state (RB*) by oxygen from air generates the superoxide anion radical and RB radical cation (RB•+), which further interacts with diarylphosphine oxide to provide the radical cation and ground-state RB to complete the photoredox cycle. Cinnamic acid could not be oxidized by RB*, owing to the high oxidation potential (E1/2ox = +2.01 V vs. SCE in MeCN).
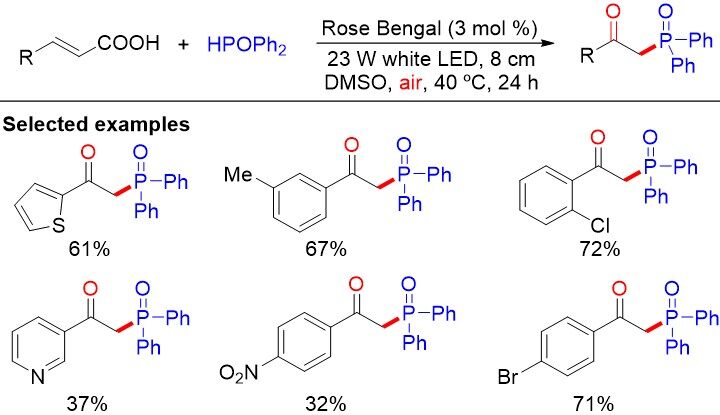
Figure 35. Visible-light photoredox-catalyzed transition metal-free cross-coupling reaction of alkenyl carboxylic acids with diarylphosphine oxides.
Later, Jung and Kim[61] introduced a visible-light photoredox-catalyzed phosphorylation of vinyl azides to prepare β-ketophosphine oxides under mild reaction conditions. This represents a convenient method by utilizing inexpensive and commercially available eosin Y as a photocatalyst and avoiding additives, oxidants and metals [Figure 36]. Among the various organic solvents, dichloromethane was found to be a useful support for the reaction and H2O could also promote the process smoothly as a benign and green solvent. With regards to the aromatic moiety of the vinyl azides, a variety of electron-deficient and electron-rich substrates could easily take part in the reaction under optimized reaction conditions and generate the desired β-ketophosphine oxides in good to excellent yields. To further certificate the reaction mechanism, the addition of excess 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) to the reaction under standard conditions resulted in a trace amount of the corresponding β-ketophosphine oxide, supporting a radical pathway in the present reaction. The mechanistic course for this reaction is displayed in Figure 36. The photocatalyst eosin Y reaches its excited state, eosin Y*, under visible-light irradiation. Initially, phosphinous acid is oxidized by eosin Y* via a SET process to generate the radical cation. The phosphinoyl radical is then formed upon deprotonation and undergoes the following radical addition with vinyl azides to afford the iminyl radical intermediate, which is reduced by eosin Y•- to furnish the corresponding anion. Eventually, protonation and hydrolysis lead to the generation of the β-ketophosphine oxides.
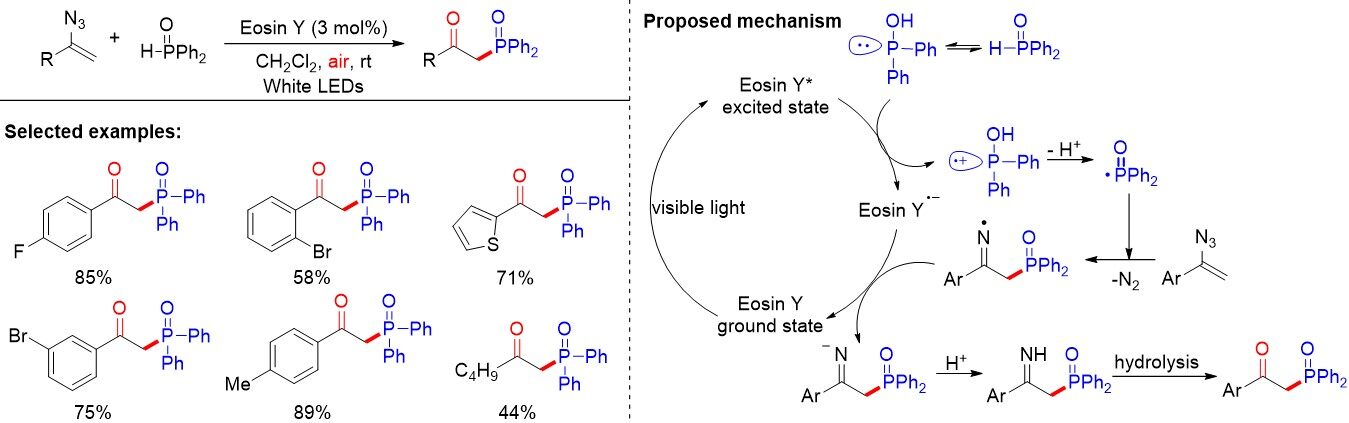
Figure 36. Visible light-mediated photocatalytic phosphorylation of vinyl azides as a mild synthetic route to β-ketophosphine oxides.
On the basis of previous reports of the hydroxyphosphorylation of alkenes[62,63], Yi et al.[64] furnished the copper-catalyzed direct hydroxyphosphorylation of electron-deficient alkenes with H-phosphine oxides and dioxygen, delivering various β-hydroxyphosphine oxides bearing a quaternary carbon center in moderate to good yields [Figure 37]. The authors proposed that the reduction of the resulting hydroperoxide by HP(O)Ph2 was the key step of the reaction.
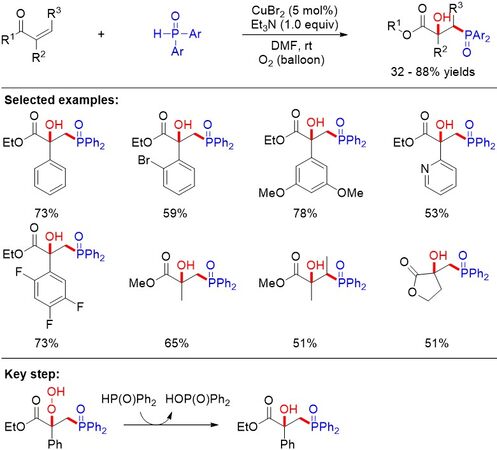
Figure 37. Hydroxyphosphorylation and oxyphosphorylation of electron-deficient alkenes.
Peroxide-containing compounds have attracted considerable attention in pharmacy, biochemistry and food chemistry. Due to the low dissociation energy of the O-O bond of the peroxy group, the synthesis of organic peroxides is particularly challenging. After disclosing an iron-catalyzed three-component carbonylation-peroxidation reaction of styrenes, aldehydes and hydroperoxides, Chen et al.[65] provided a direct approach to access multi-substituted β-phosphoryl peroxides by the reaction of alkenes, H-P(O) compounds and tert-butyl hydroperoxide (TBHP) [Figure 38]. It is noteworthy that the reaction efficiency shows the opposite trend to the stability of the P-center radical generated in situ by the SET oxidation of the organophosphorus precursors following the order: (EtO)2P(O)-H > (EtO)PhP(O)-H ≫ Ph2P(O)-H. Unfortunately, diarylphosphine oxides were not tolerated in the reaction. Moreover, the obtained β-phosphoryl peroxides could be easily transformed into β-phosphoryl epoxides with moderate diastereoselectivity using lithium bis(trimethylsilylsilyl)amide as the base.
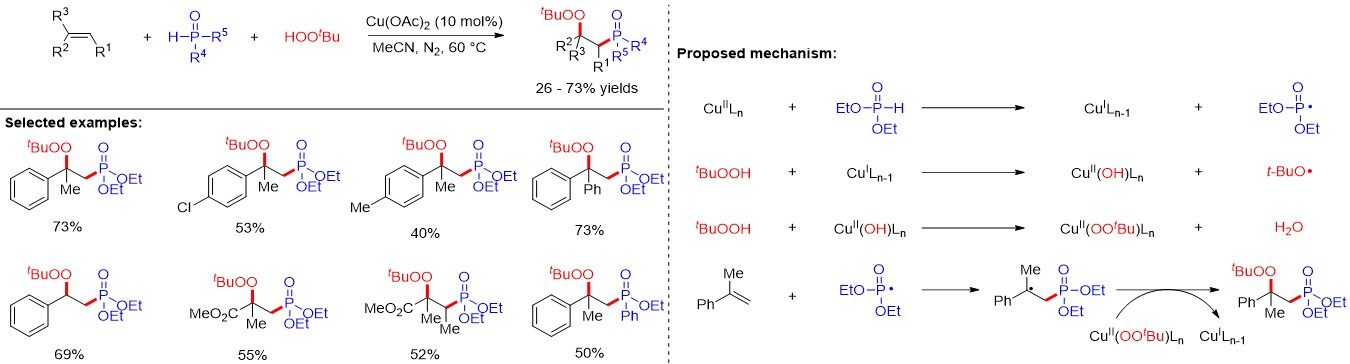
Figure 38. Copper-catalyzed three-component phosphorylation-peroxidation of alkenes.
In 2019, Shen et al.[66] established a system to achieve the P(O)-radical-mediated difunctionalization of alkenes with diarylphosphine oxide and peroxides by employing a low-cost cobalt (II) catalyst. This afforded a wide variety of phosphonation-peroxidation products in a one-pot manner [Figure 39]. The authors proposed that tert-butyloxy and tert-butylperoxy radicals were generated from excess tert-butyl hydroperoxide via a Co(III)/Co(II) catalytic cycle. The tert-butyloxy radical then abstracted the hydrogen atom of diarylphosphine oxide to produce the phosphorus radical, which was different from the generation of the P-centered radical through SET oxidation. After the addition of the phosphorus radical to the alkenes, the carbon radical undergoes a radical-radical cross-coupling with a tert-butylperoxy radical to afford the final product.
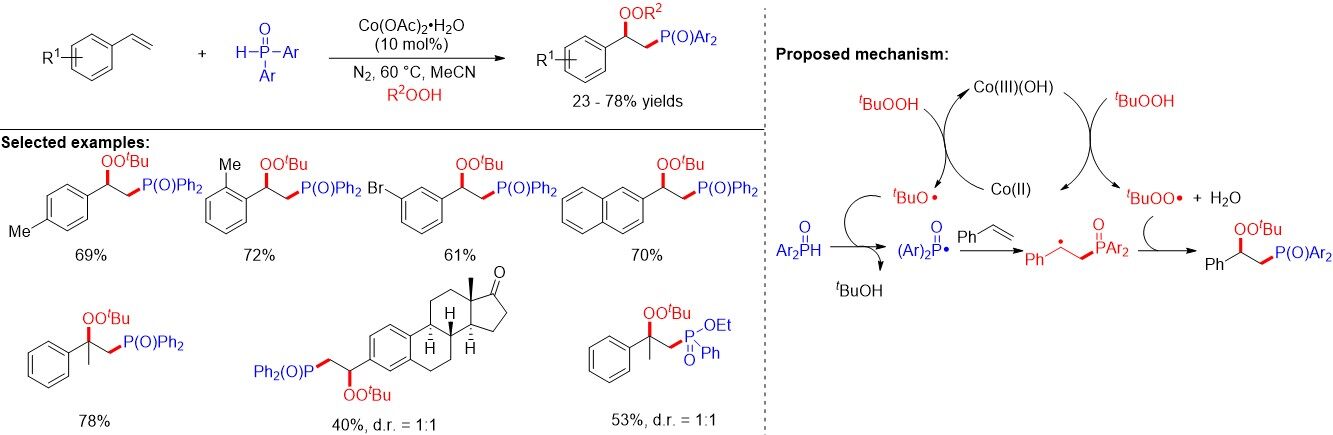
Figure 39. Co(II)-catalyzed difunctionalization of alkenes with diarylphosphine oxide and peroxide.
As a variation of the above-mentioned radical coupling mechanism, the stoichiometric metal-mediated sequential oxidation of carbon radicals to carbocations ending with nucleophilic attack has also become a powerful tool for the oxyphosphorylation of alkenes. For example, in 2015, Zhou et al.[67] developed a
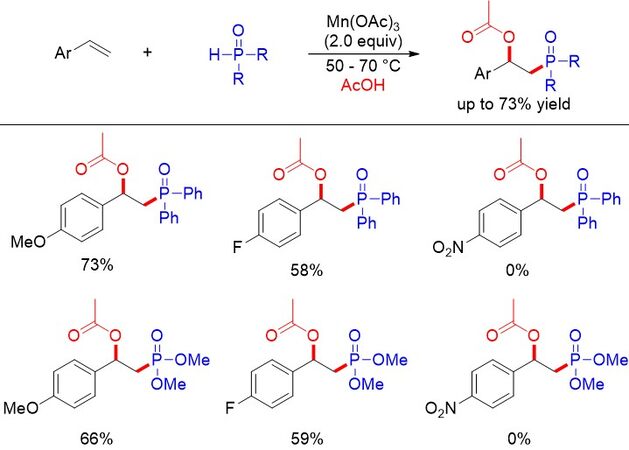
Figure 40. Mn(II)-mediated acetoxyphosphorylation of styrenes.
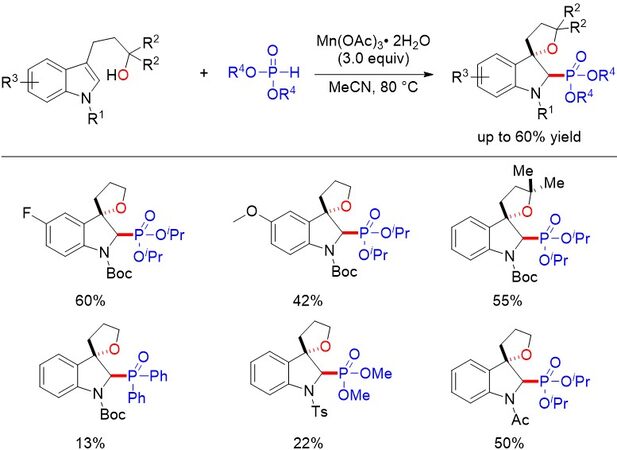
Figure 41. Mn(III)-mediated synthesis of 3,3-spirocyclic 2-phosphonoindolines via a dearomative addition of phosphonyl radicals to indoles.
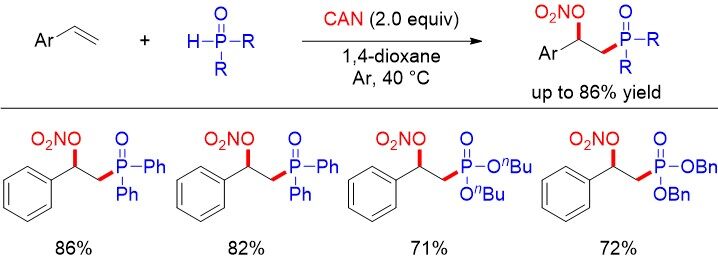
Figure 42. Ce(IV)-promoted phosphinoylation-nitratation of alkenes.
Difunctionalization with C-N/C-S/C-F bond formation
The unique properties of fluoride, as well as the significant application of 18F-labelled organic compounds as contrast agents for positron emission tomography, has attracted significant attention from chemists for the development of new methods for the construction of C-F bonds under mild reaction conditions. Moreover, fluorinated phosphonates have also found wide applications, especially in biomedical studies. However, methods for the synthesis of the important class of β-fluorinated phosphonates are extremely limited. In 2013, Zhang et al.[70] reported a mild and catalytic method for the phosphonofluorination of unactivated alkenes with AgNO3 as a catalyst [Figure 43]. This protocol features good stereoselectivity, a broad substrate scope and wide functional compatibility. The authors proposed that silver-catalyzed oxidative generation of phosphonyl radicals and silver-assisted fluoride atom transfer were involved in the reaction.
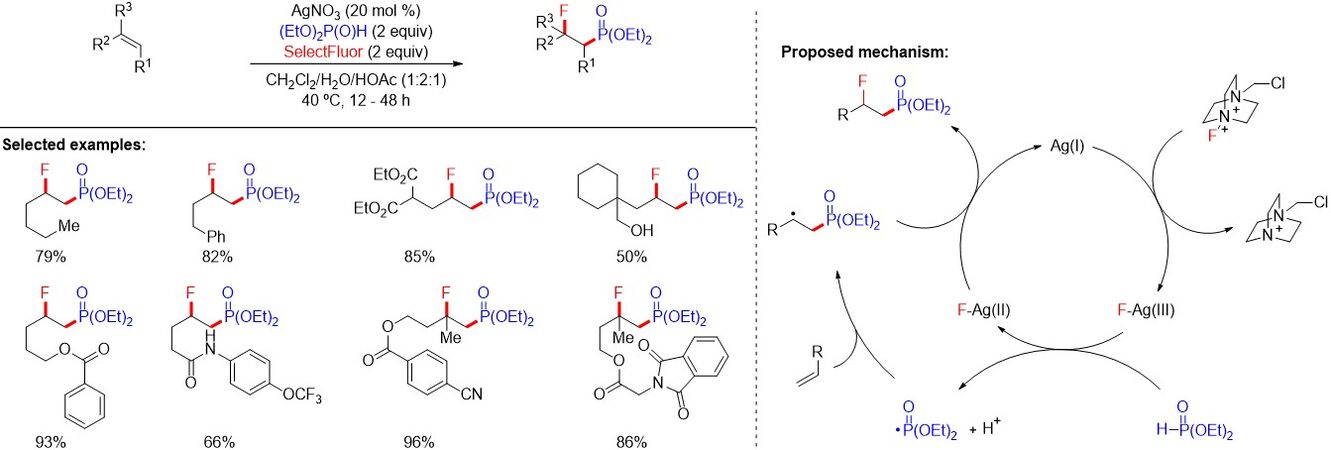
Figure 43. Silver-catalyzed radical phosphonofluorination of unactivated alkenes.
Nitrogen heterocyclic components have been found abundant applications as important structural motifs in biologically active compounds. The difunctionalization of 2-allylaniline derivatives represents a direct and atom-economical approach for the preparation of indolines with different substituents. However, the synthesis of phosphorylated indoline by aminophosphophination via a radical cascade cyclization has never been realized. In 2015, Zhang et al.[24] described a copper-catalyzed radical cascade cyclization for the synthesis of phosphorylated indolines [Figure 44]. This reaction features mild conditions with low-cost copper salts as catalysts. Notably, the authors proposed that an intramolecular Ullmann-type reaction, followed by reductive elimination to release the product, was involved in the reaction.
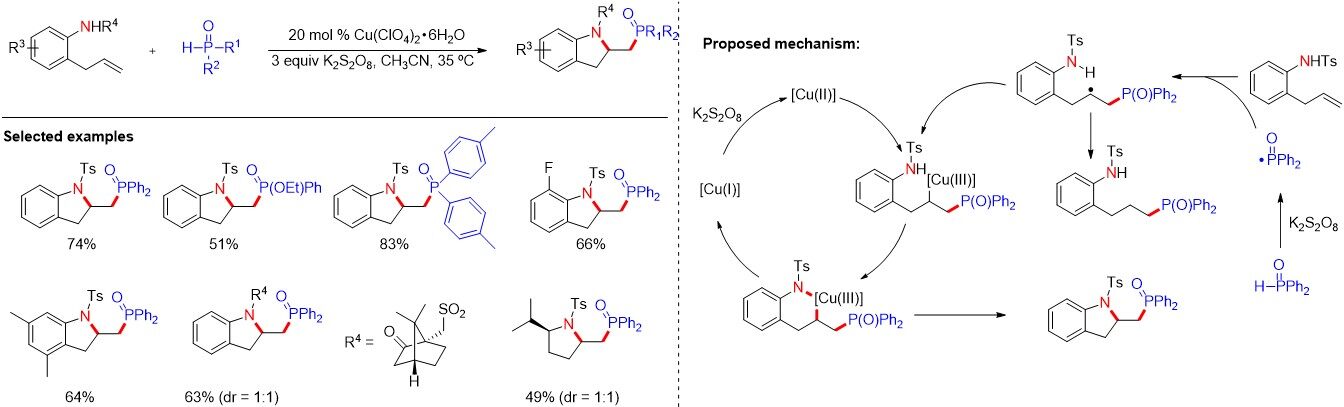
Figure 44. Copper-catalyzed radical cascade cyclization for the synthesis of phosphorylated indolines.
Although bifunctional α,β-aminophosphinoyl compounds possess special biological activities, their synthetic methods are limited. In contrast with the work of Yang, in 2017, Li et al.[71] reported a phosphinoyl radical-initiated intermolecular α,β-aminophosphinoylation of alkenes [Figure 45]. This one-pot three-component reaction is performed under mild conditions to afford α,β-aminophosphinoylation products. In addition to diarylphosphine oxides, dialkylphosphites are also tolerated in the reaction. As illustrated in Figure 45, the carbon radical coordinating with the Cu(II)-aniline complex delivers the key Cu(III)-complex, which undergoes a reductive elimination step to give the final product. Similar work using FeCl3 as the catalyst and TBHP as the oxidant was described by Wang et al.[72] in 2018.
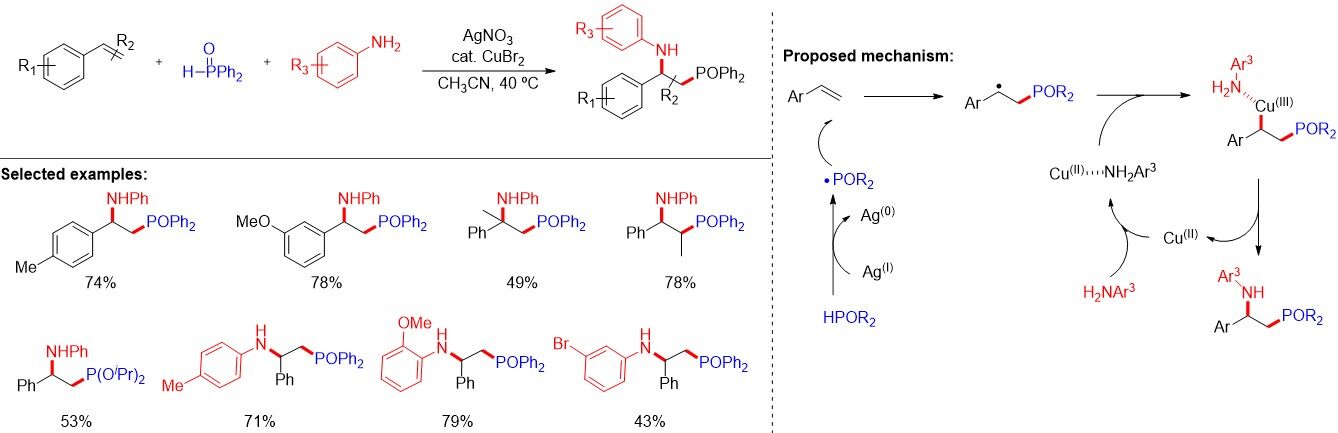
Figure 45. Phosphinoyl radical-initiated α,β-aminophosphinoylation of alkenes.
Compounds containing nitrogen and phosphorus atoms have been widely applied in many research areas, and the phosphinoylazidation of alkenes serves as a direct method for the construction of nitrogen- and phosphorus-containing compounds. Although phosphinoylazidation was realized[73] in 2015 using a stoichiometric amount of the oxidative radical initiator Mn(OAc)3•H2O, the catalytic phosphinoylazidation of alkenes remains unresolved. Recently, Ma et al.[74] developed an iron phthalocyanine-catalyzed radical phosphinoylazidation of alkenes for the facile synthesis of β-azido-phosphine oxide under relatively mild reaction conditions [Figure 46]. Mechanistic studies revealed an unusually low activation energy of 4.8 kcal/moL for the azido transfer process from the azidyl iron(III) phthalocyanine species (PcFeIIIN3) to a benzylic radical.
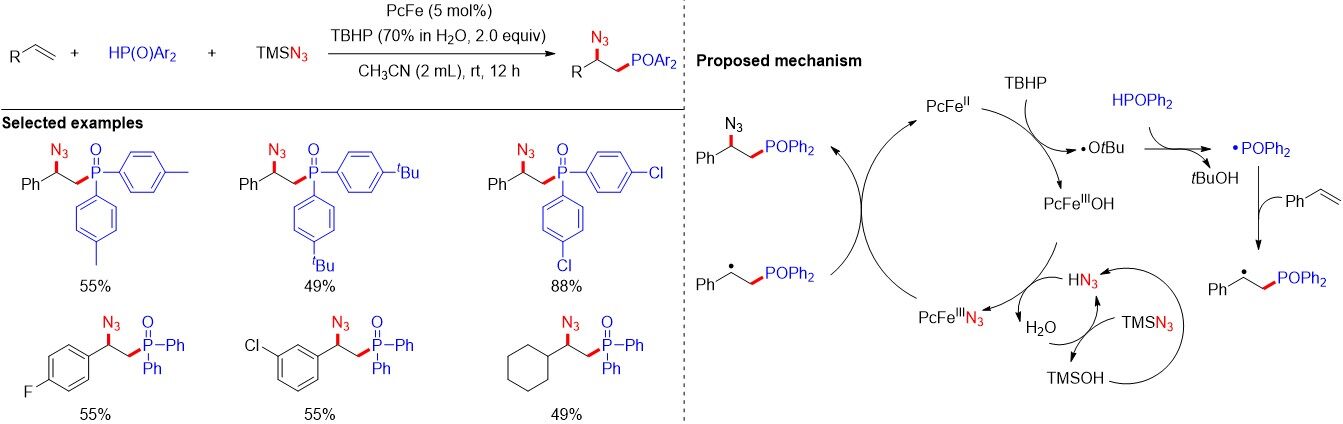
Figure 46. PcFe-catalyzed phosphinoylazidation of alkenes.
Both phosphinoyl and thiocyano are valuable functional groups commonly found in pharmaceutically active molecules. Thiocyanodiphenylphosphinoylated compounds could be applied in the development of new bioactive compounds and phosphine-containing ligands. In 2018, Tao et al.[75] established the phosphinoyl radical-initiated 1,2-bifunctional thiocyanodiphenylphosphinoylation of alkenes using CuBr2 as a catalyst and Mn(OAc)3 as an oxidant, affording a range of thiocyanodiphenylphosphinoylated compounds in reasonable yields [Figure 47]. Mechanistic studies reveal that copper-catalyzed thiocyanation is involved in the reaction.
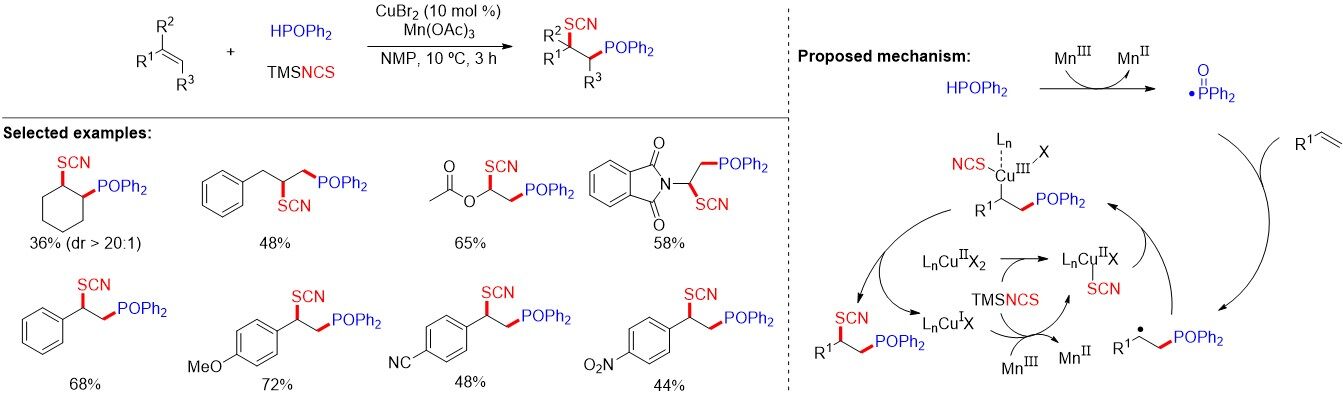
Figure 47. Phosphinoyl radical-initiated 1,2-bifunctional thiocyanodiphenylphosphinoylation of alkenes.
PHOSPHORYLATION OF ALKYNES
Hydrophosphorylation of alkynes
Alkenylphosphoryl compounds are highly useful organophosphorus compounds in organic chemistry. Compared with the transition metal-catalyzed hydrophosphorylation of alkynes, radical addition to alkynes has directly become an alternative approach to assemble functionalized alkenes. In 2018, Huang et al.[76] reported the radical hydrophosphorylation of alkynes with P(O)-H compounds, obtaining anti-Markovnikov alkenylphosphine oxides in moderate yields [Figure 48]. In particular, propargyl alcohols, which are not compatible with palladium-catalyzed hydrophosphorylation, can also deliver the desired products in good yields. This green approach avoids the use of a metal catalyst, which can serve as a complementary reaction to the metal-mediated hydrophosphorylation.
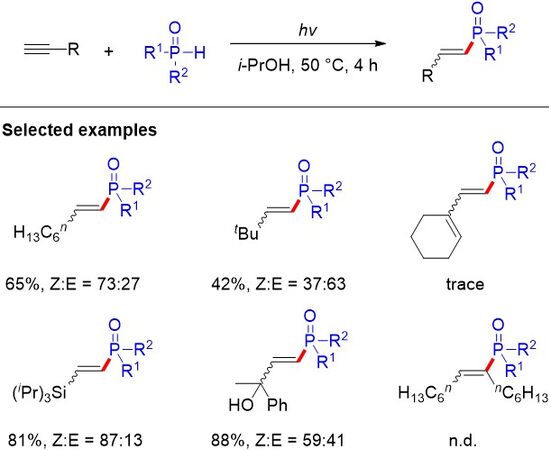
Figure 48. Radical hydrophosphorylation of alkynes with R2P(O)H generating alkenylphosphine oxides.
Radical addition to alkynes offers a straightforward, atom-economical and environmentally benign method for the synthesis of functionalized alkenes. However, the control of the stereo- and regioselectivity remains highly challenging, especially for the synthesis of Z-alkenes. In 2018, the addition of diaryl phosphine oxides to alkynes with excellent and challenging Z-stereo- and regioselectivity via photoredox catalysis was revealed by Wang et al.[77] [Figure 49]. Various terminal and internal alkynes could be transformed to higher-energy Z-alkenes. A series of mechanistic studies advocate that K2CO3, as an aqueous base, plays a decisive role in the transformation. The π-π stacking interaction with the phosphorus substituents greatly promotes Z-selectivity and the reaction proceeds through free-radical addition. A tentative mechanism for this Z-selective protocol is proposed in Figure 49. First, a phosphinoyl radical is generated via PCET oxidation in the presence of photoexcited catalysts. A phosphinoyl radical then combines with phenylacetylene to afford the α-alkenyl carbon radical. Afterwards, the SET between PC•- and the linear radical delivers the Z-alkenyl anion. In this process, Z-selectivity could be greatly improved by a proposed π-π stacking interaction between the phenyl ring of the phenylacetylene and one of the aromatic rings of diphenylphosphine oxide. Notably, an X-ray single-crystal structure showed that one of the aromatic rings of diphenylphosphine oxide and the pyridine are on the same side, suggesting the existence of the π-π stacking interaction between the aromatic rings. Finally, the Z-alkenyl anion undergoes protonation to afford Z-alkenylphosphine oxides.
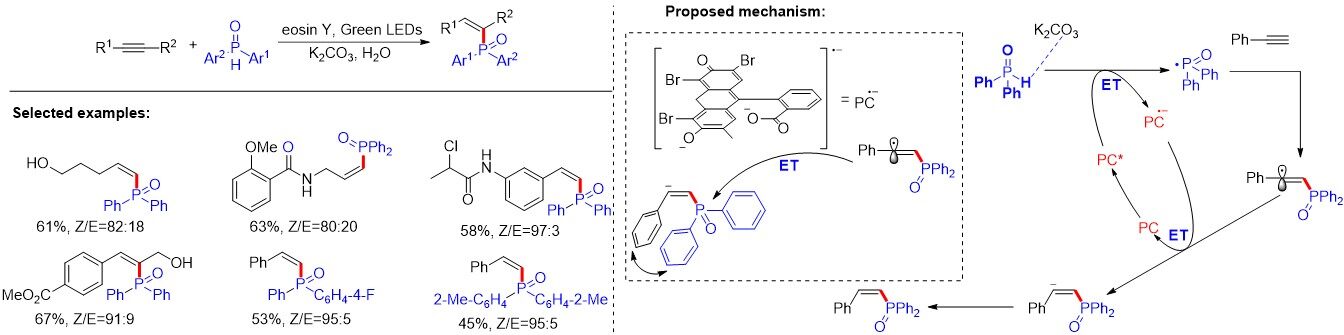
Figure 49. Z-Selective addition of diaryl phosphine oxides to alkynes via photoredox catalysis.
In contrast to the high Z-selectivity of the work of Wang et al.[77], Liu et al.[78] discovered that earth-abundant cobaloxime could convert H-phosphine oxide to its reactive radical species under visible-light irradiation. The P-centered radicals react with alkynes to generate E-alkenylphosphine oxides with excellent chemo- and stereoselectivity [Figure 50]. Linear E-alkenylphosphine oxide could be obtained from the reaction with terminal alkynes, while addition with internal alkynes leads to cyclic benzophosphine oxides and hydrogen. Notably, the mechanistic investigation shows the dual roles of cobaloxime, both as the visible-light absorber to activate the P(O)-H bond, as well as a HAT reagent. The proposed mechanism is as follows. The P(O)-H bond was activated by cobaloxime under photoexcitation to generate the phosphinoyl-centered radical. Next, the addition of the phosphinoyl radical intermediate to an alkyne gives the alkenyl carbon radical, which is captured by the Co(II) intermediate to form the vinyl Co(III) intermediate. The final protonation forms a hydrophosphorylation product and regenerates the initial Co(III) complex, which closes the catalytic cycle. In terms of internal alkynes, the alkenyl carbon radical attacks the arenes to give a cyclic radical intermediate that reacts with the Co(II) complex to give a Co(III)-H complex via HAT. In the absence of an external oxidant, the combination of cobaloxime [Co(III)-H] and an electron result in the release of the captured proton and electron as hydrogen (detected by GC).
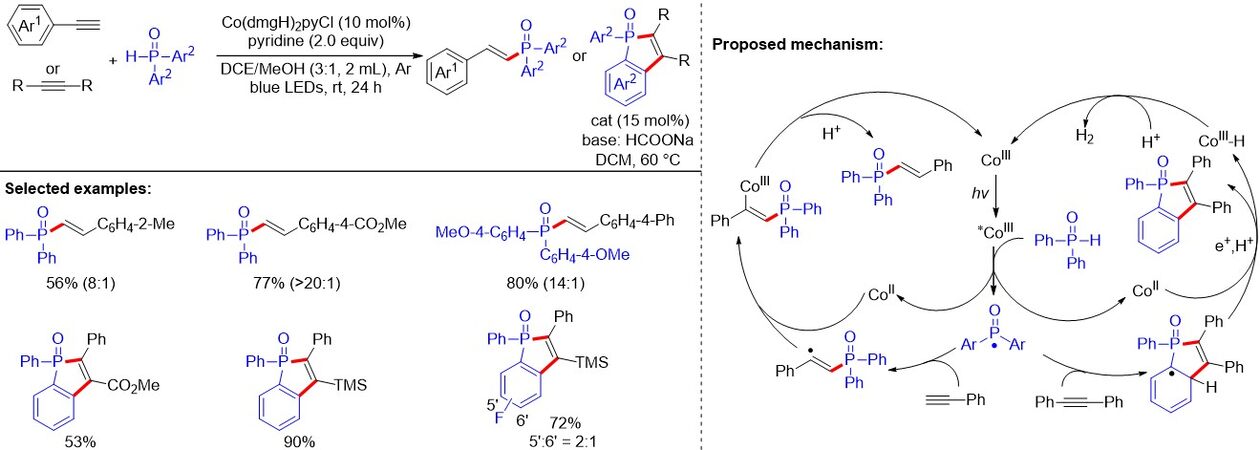
Figure 50. Cobaloxime catalysis-based selective synthesis of alkenylphosphine oxides under visible light.
Different from the transition metal or photoredox-mediated generation of P-centered radicals through the oxidative process via SET, recently, Hou et al.[79] introduced a stereo- and regioselective cis-hydrophosphorylation reaction of the internal alkyne of 1,3-enynes via reactive Ni(II)-phosphorus species migratory insertion into the internal alkyne, giving rise to a variety of 1,3-dienes with good yields [Figure 51]. Notably, the phosphorus radical derives from the hydrogen atom abstraction of the H-P(O) compound. In the reaction, the reactive Ni(II)-phosphorus species is generated from visible-light irradiation of the NiCl2(PPh3)2 and the function of the ligand should not be neglected. A possible mechanism for this double-component transformation involving visible light-induced nickel phosphine species generation via an Ni(II)/Ni(I)/Ni(II) cycle was proposed. The coordination of the Ni(PPh3)2Cl2 with 1,3-enyne forms the Ni(II) intermediate, which generates the Ni(I) species and the chlorine atom radical Cl• via the LMCT process under visible-light irradiation. The phosphorus atom radical, in situ generated through hydrogen atom abstraction by Cl•, could re-bond with Ni(I) species, resulting in the formation of Cl-Ni(II)-P(O)Ph2 species. The insertion of the Cl-Ni(II)-P(O)Ph2 species into the C≡C bond of the 1,3-enyne generates the intermediate alkenyl Ni(II) intermediate. Finally, the rapid protonation of the intermediate by MeOH produces the desired cis-hydrophosphorylation product.
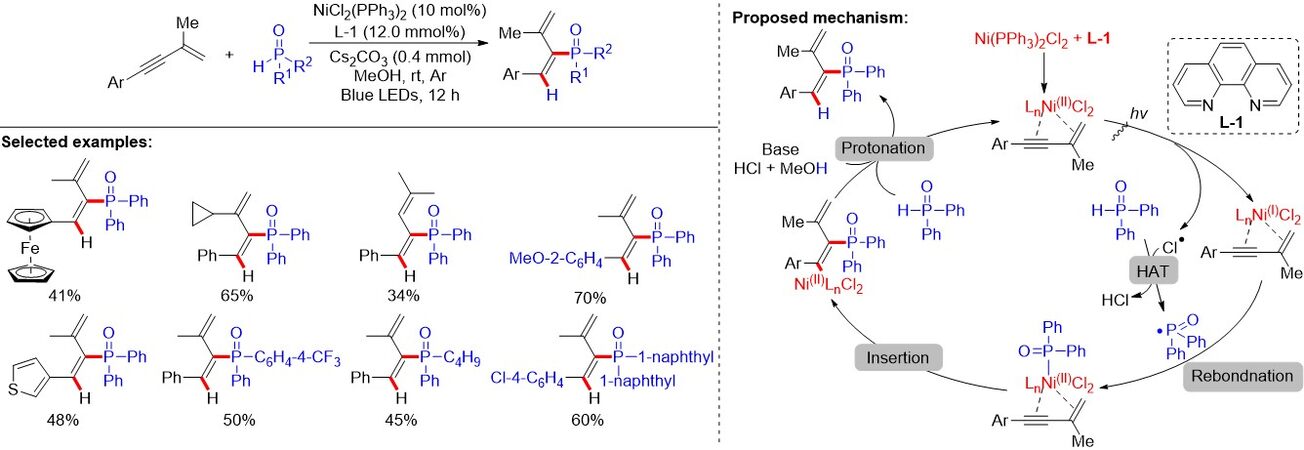
Figure 51. Stereo- and regioselective cis-hydrophosphorylation of 1,3-enynes enabled by visible-light irradiation of NiCl2(PPh3)2.
Difunctionalized phosphorylation of alkynes
A series of transition metal-catalyzed aerobic phosphorylation reactions of alkynes or alkynylcarboxylic acids have been reported by Zhang et al.[80], Chen et al.[81], Yi et al.[82] and Zhou et al.[83], delivering synthetically and biologically valuable β-ketophosphonates. In 2018, Zhong et al.[84] reported a simple and efficient method for the oxyphosphorylation of alkynes using LiOH without the assistance of any transition metal, delivering a wide range of β-ketophosphonates. Mechanistic studies indicate that the inorganic base promoting the oxyphosphorylation and H2O plays a synergistic role in the formation of the products [Figure 52].
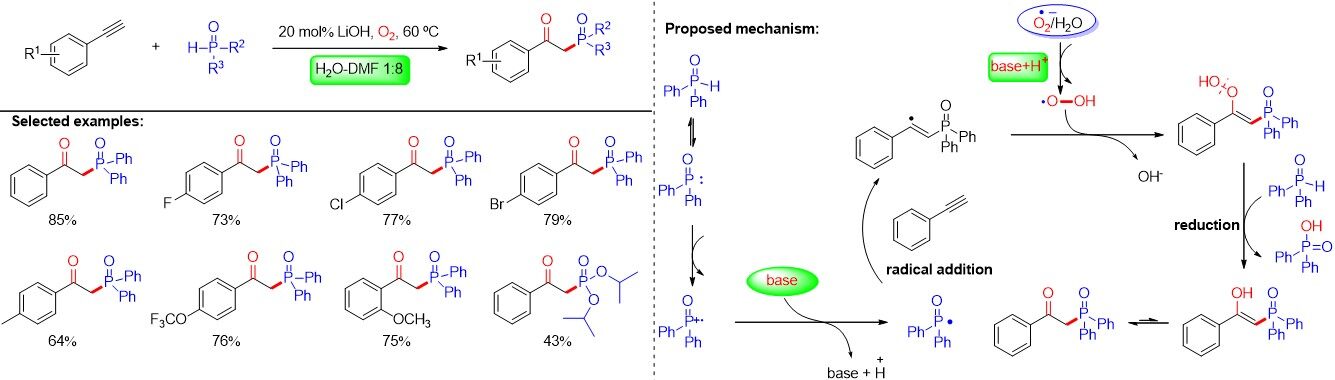
Figure 52. Base-promoted direct oxyphosphorylation of alkynes with H-phosphine oxides in the presence of water.
Recently, Tao et al.[85] described a copper-catalyzed difunctional cyano-, thiocyano- and chlorophosphorylation reaction of alkynes with P(O)-H compounds and various coupling partners (TBACN, TMSNCS and TMSCl) [Figure 53]. A diverse array of tri- and tetra-substituted alkenyl phosphine oxides/phosphonates was constructed regio- and stereoselectively. Mechanistic studies indicate that the reaction is initiated by the addition of a P-centered radical to the alkyne to form an alkenyl radical, which could combine with Cu(II) specie to form a Cu(III) complex. A reductive elimination step then leads to the formation of the final cyano-, thiocyano- or chlorophosphoryl alkenes, which represent valuable building blocks with potential medicinal, agrochemical and functional material applications.
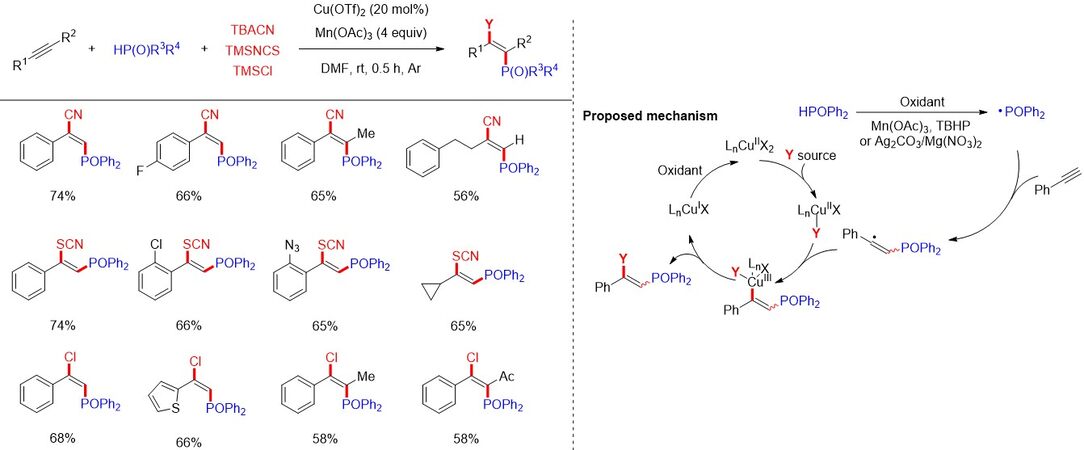
Figure 53. Difunctional cyano-, thiocyano- and chlorophosphorylation reaction of alkynes with P(O)-H compounds.
Radical cascade cyclization phosphorylation of alkynes
π-Conjugated phosphole molecules have become vitally important in the blooming area of organic light-emitting diodes and photovoltaic cells. Transition metal-mediated synthesis of benzo[b]phosphole oxides have been reported, for instance, Zhang et al.[86] described a copper-catalyzed cycloaddition between secondary phosphine oxides and alkyne to give benzophosphole oxides. However, it is still highly appealing to develop an approach for the construction of benzo[b]phosphole oxides without the requirement for expensive transition metal catalysts or toxic reagents. In 2016, Quint et al.[87] described a metal-free visible-light photoredox-catalyzed synthesis of benzo[b]phosphole oxides using N-ethoxy-2-methylpyridinium tetrafluoroborate as the oxidant and eosin Y as the catalyst under mild conditions [Figure 54]. Alkynes bearing alkyl, ester and alcohol groups could be transformed to the desired products. However, the aryl migration of the diphenylphosphine oxides, including electron donor or electron acceptor groups at the aromatic rings, results in the formation of regioisomers of benzophospholes.
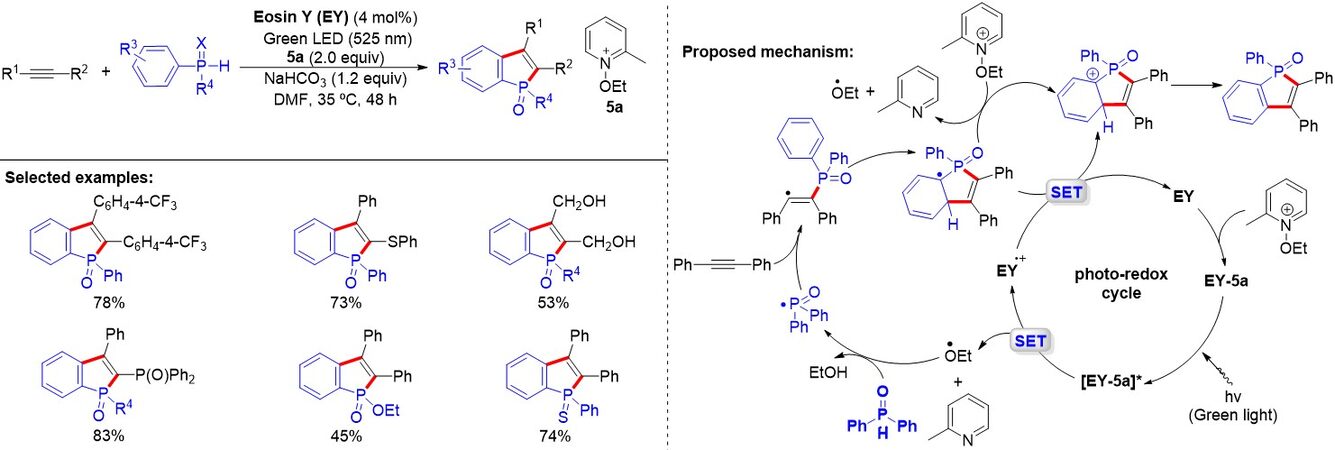
Figure 54. Synthetic and mechanistic investigations of metal-free visible light-photocatalyzed synthesis of benzo[b]phosphole oxides.
Mechanistic investigations show the formation of a ground-state electron donor-acceptor complex (EDA) between the pyridinium salt and eosin Y. A plausible mechanism for the generation of the product is given in Figure 54. The formation of the EDA complex between the eosin Y and the onium salt initiates the reaction. Upon absorption of a green photon and a SET event, the EDA complex generates the unstable ethoxy radical, which abstracts a hydrogen from the secondary phosphine oxide to rapidly deliver the corresponding phosphinoyl radical. Next, this radical adds to the alkyne to give the alkenyl radical. Subsequently, such a radical was trapped by the phenyl ring of the phosphine oxide to produce the cyclohexadienyl radical, which (with a redox potential of about -0.1 V) is readily oxidized by EY•+ [E0(EY•+/EY) = 0.70 V/SCE] to release the photocatalyst and provide the Wheland intermediate. Finally, the Wheland intermediate is immediately rearomatized to yield the desired benzophosphole oxide upon deprotonation with NaHCO3.
Heterocyclic skeletons, which are ubiquitous in natural products and exhibit a wide range of pharmacological properties, have attracted significant attention for the synthesis of pharmaceutically relevant molecules. Furthermore, the intermolecular radical addition/cyclization cascade reaction represents an efficient and powerful strategy to forge highly functionalized organic heterocycles. Although significant progress has been achieved, the elaboration of the phosphorylation products of alkynes remains highly desirable. In 2017, Zhu et al.[88] reported a silver-mediated radical 5-exo-dig cyclization of 2-alkynylbenzonitrile with disubstituted phosphine oxides and H2O for the synthesis of phosphinylated 1-indenones [Figure 55]. Multiple bonds, including C-P, C-C and C-O bonds, were successively constructed under atmospheric conditions. The reaction displays flexible structural modification, a broad substrate scope and high functional group tolerance. Furthermore, P-centered radical addition, 5-exo-dig cyclization and hydrolysis processes were proposed as the key steps in the reaction by the authors.
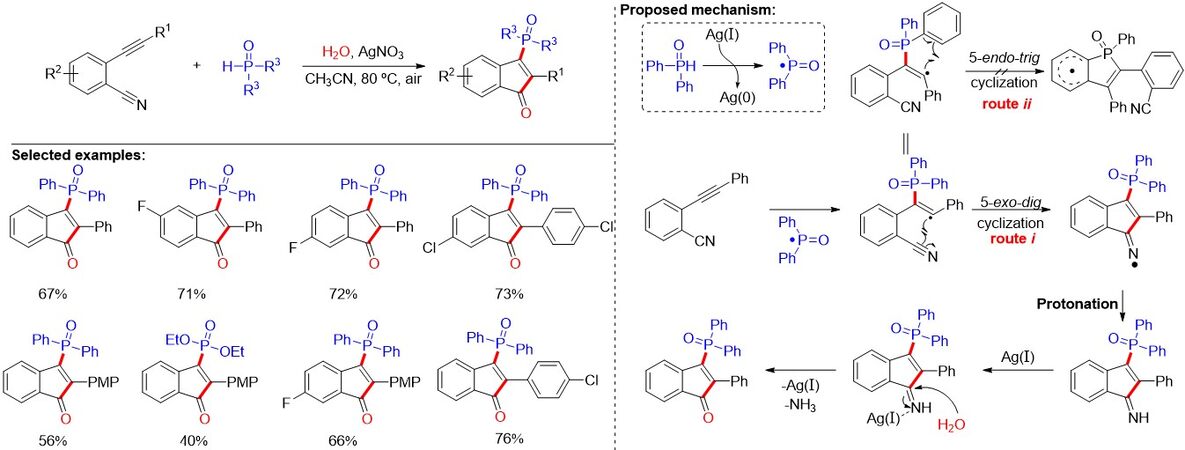
Figure 55. Silver-mediated radical 5-exo-dig cyclization of 2-alkynylbenzonitrile with H-P(O) compounds.
In 2020, Hou et al.[89] described intermolecular hydrophosphorylative cyclizations of enynes and dienes to access heterocycles under visible-light irradiation [Figure 56]. The phosphinoyl radicals were formed via direct HAT between the phosphine oxides and photoexcited catalyst. The desired heterocycles were obtained in good yields via the radical addition/cyclization/HAT reaction sequences of 1,6-enynes and 1,6-dienes.
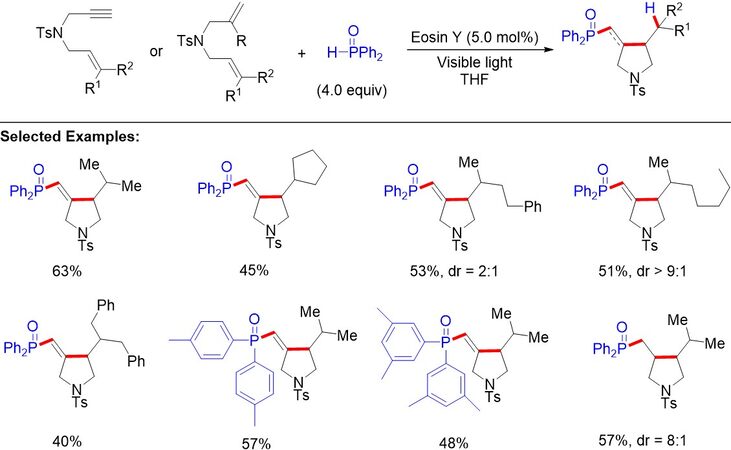
Figure 56. Visible light-mediated hydrophosphorylative cyclizations of enynes and dienes.
Phosphorylated 2-oxazolidinones have found numerous applications in pharmaceutical chemistry, homogeneous catalysis and organic materials. In 2020, Huang et al.[90] described an efficient and selective protocol for the straightforward synthesis of various 5-[(diarylphosphoryl)methyl]oxazolidin-2-ones via the copper-catalyzed difunctionalization of the propargylic amines with CO2 and phosphine oxides [Figure 57]. The approach exhibits a wide substrate scope, excellent functional group compatibility and exclusive selectivity. Mechanistic studies reveal that a one-pot tandem cyclization/radical addition sequence, along with the phosphorylation/cyclization scheme, is involved in the reaction.
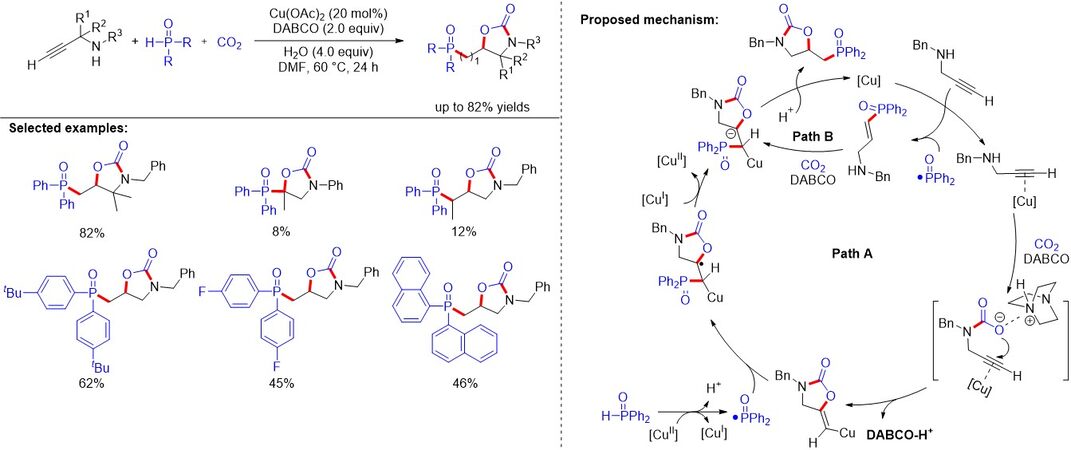
Figure 57. Copper-catalyzed phosphonocarboxylative cyclization reaction of propargylic amines.
Thioflavones are valuable functional motifs found in numerous natural products, drugs and materials. The incorporation of phosphorus functional groups into the skeleton of thioflavones has attracted extensive attention. In 2019, a visible light-induced metal-free phosphorylation of methylthiolated alkynones to synthesize 2-phosphorylated thioflavones was developed by Liu et al.[91] [Figure 58]. Interestingly, the non-toxic, low cost and easily available water acts as the reaction medium to promote this reaction and this is the first example of 4CzIPN as a photocatalyst to construct C-P bonds in water. To demonstrate the applicability of the approach, they carried out a gram-scale synthesis and obtained the product in good yield (67%). A plausible reaction mechanism for this reaction is shown in Figure 58. Initially, the photocatalyst 4CzIPN is activated to its excited-state 4CzIPN* under the irradiation of visible light. Diphenylphosphine oxide is usually in equilibrium between tri- and tetra-coordinated forms. A SET between the tri-coordinated form of diphenylphosphine oxide and 4CzIPN* then leads to the phosphoryl radical and radical anion 4CzIPN•-. Subsequently, addition of the phosphoryl radical to the triple bond and the following intramolecular cyclization furnishes the final product and releases the methyl radical. Moreover, the photoredox cycle is completed after the oxidation of the radical anion 4CzIPN•- by dilauroyl peroxide.
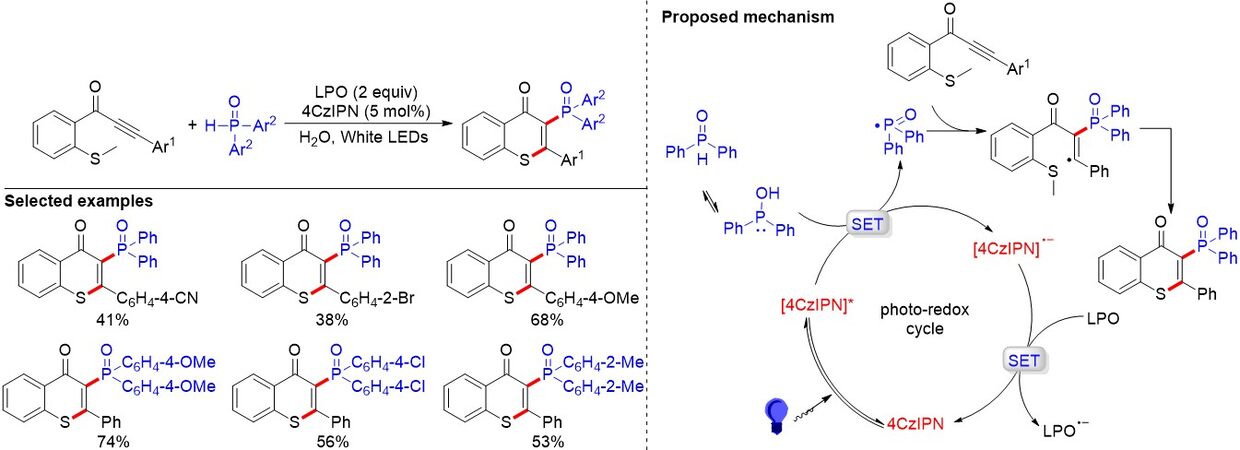
Figure 58. Visible light-induced metal-free synthesis of 2-phosphorylated thioflavones in water.
PHOSPHORYLATION OF ISOCYANIDES
Arylphenylisonitriles, which contain a C≡N π-bond, are good radical acceptors and can be employed to construct diverse 6-substituted phenanthridines. In spite of the development of the silver-promoted radical phosphorylation of isonitriles, it is still highly desirable to exploit a more effective and environmentally friendly protocol. In 2016, an efficient visible light photoredox-catalyzed tandem phosphorylation/cyclization reaction of three kinds of radical acceptors, including 2-arylphenylisonitrile, vinyl isocyanides and N-arylacrylamides, with diphenylphosphine oxide, was unmasked by Li et al.[92] [Figure 59]. This reaction generates phosphorus-containing phenanthridines, isoquinolines and indolin-2-ones by the construction of both the C-P and C-C bonds with [Ir(ppy)2(dtbpy)]PF6 as the catalyst, K2S2O8 as the oxidant and CsF or Cs2CO3 as the base. Phosphine oxides, such as tertbutyl(phenyl)phosphine oxides and ethyl pheylphosphinates, and a series of vinyl isocyanides could also be tolerated in this transformation. It is remarkable that this strategy can be utilized for the synthesis of bioactive oxindoles using N-arylacrylamide as a radical receptor.
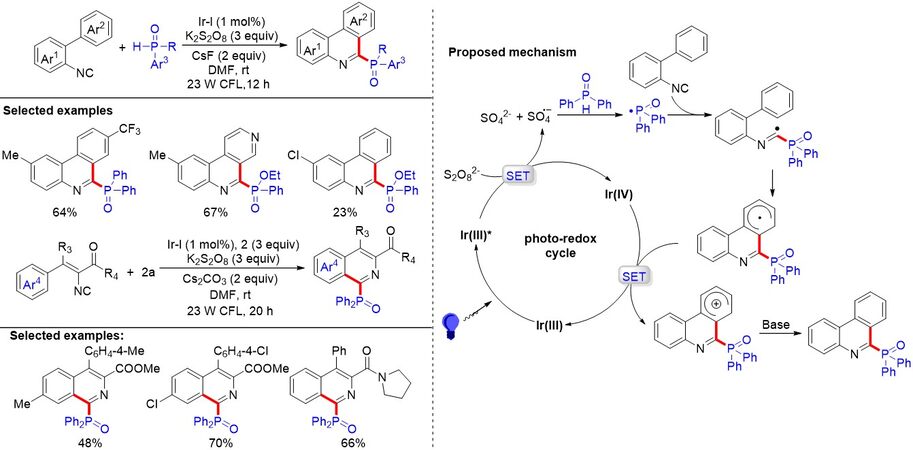
Figure 59. Visible light-induced cascade reaction of isocyanides and N-arylacrylamides with diphenylphosphine oxide via radical C-P and C-C bond formation.
On the basis of mechanistic studies and the literature, a tentative mechanism is proposed in Figure 59. Firstly, the ground-state photocatalyst Ir(III) is converted into the excited-state species Ir-1(III)* under visible-light irradiation, which then reduces the persulfate anion to generate Ir(IV), a sulfate dianion and a sulfate radical anion. Next, after a HAT process between the active sulfate radical anion and diphenylphosphine oxide, the crucial intermediate P-centered radical is produced. The intermolecular radical addition of P-centered radicals to radical acceptors then results in the imine radical intermediate. It then undergoes the intramolecular cyclization to generate a cyclohexadiene-type free radical, which can be oxidized by Ir(IV) to regenerate Ir(III) and provides the cationic intermediate. Finally, in the presence of a base, the cationic intermediate undergoes deprotonation to give the desired product.
Benzothiazole and its derivatives, which are vital building blocks in synthetic intermediates, usually have unexpected biological and pharmacological properties. Previously, Yang et al.[93] disclosed a novel method to synthesize 2-phosphorylated thioethers. Nevertheless, the cascade reaction heavily relies on the use of stoichiometric metal salts and a high reaction temperature. Later, the same group discovered metal-free photoinduced radical cascade reactions of isocyanides with phosphine oxides for the formation of 2-phosphoryl benzothiazoles [Figure 60]. These cascade reactions involve concomitant C(sp3)-S bond cleavage and imidoyl C-S formation. This environmentally friendly approach was decorated with organic dye Rose Bengal as the photocatalyst, without additional transition metal or peroxide oxidants and a variety of 2-phosphoryl benzothiazoles produced in moderate to good yields.
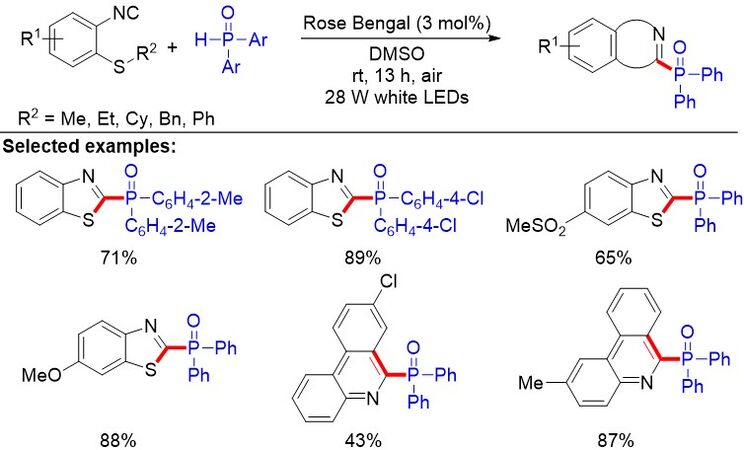
Figure 60. Metal-free photoinduced radical C-P and C-S bond formation for the synthesis of 2-phosphoryl benzothiazoles.
Despite significant progress having been achieved in the photoredox-mediated tandem cyclization of isocyanides with organic metal-based photocatalysts, such as Ir and Ru, the dye-catalyzed P-radical initiated cascade cyclization of isocyanides remains highly intriguing. In 2020, 2,4,5,6-tetrakis(3,6-di-tert-butyl-9H-carbazol-9-yl)-isophthalonitrile (4CzIPN-tBu) was utilized to realize the radical cascade cyclization reaction for the synthesis phosphorylated N-heteroaromatics using various isocyanide-containing substrates, such as 2-arylphenylisonitrile, 2-isocyanoaryl thioethers and vinyl isocyanides, via a visible light-induced PCET strategy [Figure 61][94]. A broad range of phosphorylated aromatics, including phenanthridines, quinolines and benzothiazoles, were efficiently constructed. This transition metal-free strategy should be of great interest for the future development of phosphorus radical-based reactions.
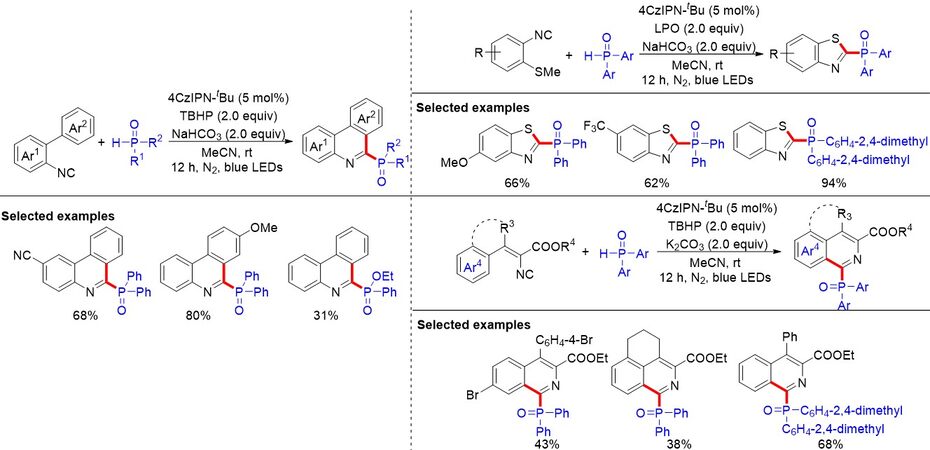
Figure 61. 4CzIPN-tBu-catalyzed proton-coupled electron transfer for photosynthesis of phosphorylated N-heteroaromatics.
CROSS-COUPLING PHOSPHORYLATION REACTIONS
Transition metal/photoredox dual catalyzed phosphorylation reactions of (pseudo)halides
Carbonyl-containing compounds and phenols, ubiquitous in diverse organic compounds, are readily available in nature and industry. In addition, the easily generated alkenyl and aryl C-O electrophiles are attractive in photoredox and transition metal dual catalysis. In 2017, Liao et al.[95] reported a phosphorylation of alkenyl and aryl C-O bonds at room temperature via photoredox/nickel dual catalysis [Figure 62]. Diverse structurally important alkenyl phosphonates and aryl phosphine oxides are generated in moderate to excellent yields. On the basis of their exploration and previous studies, they proposed the mechanism for the reaction. Firstly, the P-centered radical is generated through a SET between phosphine oxides with the excited photocatalyst following base-promoted deprotonation. Meanwhile, the oxidative addition between the tosylate and a Ni0 species gives the Ni(II) intermediate, which rapidly captures the P-centered radical to afford the Ni(III) complex. Finally, the desired product was formed through facile reductive elimination. Notably, both the photocatalyst and nickel catalyst are regenerated to complete the dual catalytic cycle via the reduction of complex LnNiI-OTs by the reduced form of a Ru photocatalyst.
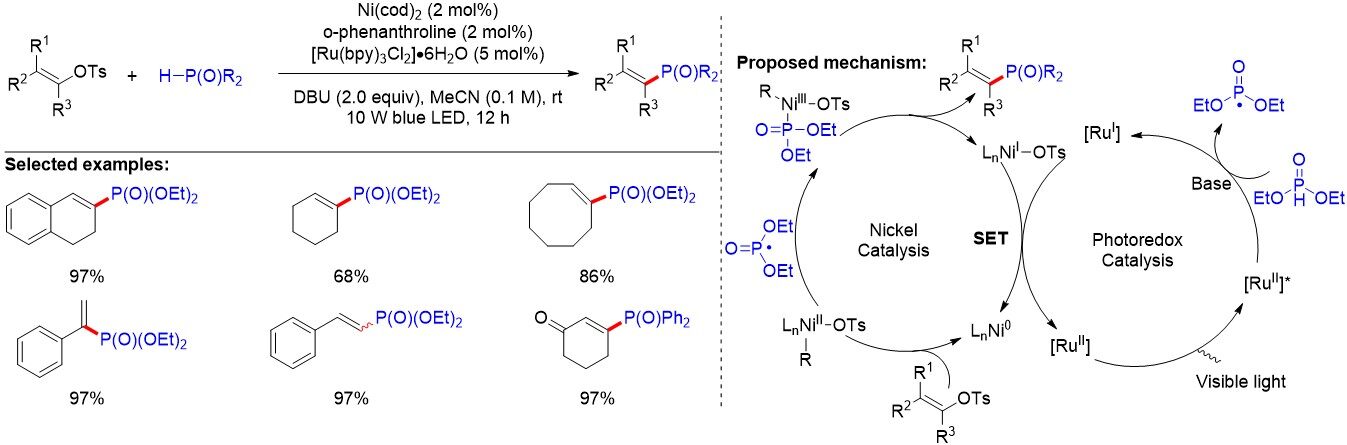
Figure 62. Photoredox/Ni dual catalyzed phosphorylation of alkenyl and aryl C-O bonds.
The strong base and/or high energy ultraviolet irradiation required for the metal-free photoredox-mediated cross-coupling phosphorylation reaction of aryl halide restricts its functionality compatibility. Therefore, the development of an efficient and mild phosphorylation reaction is urgently required. In 2021,
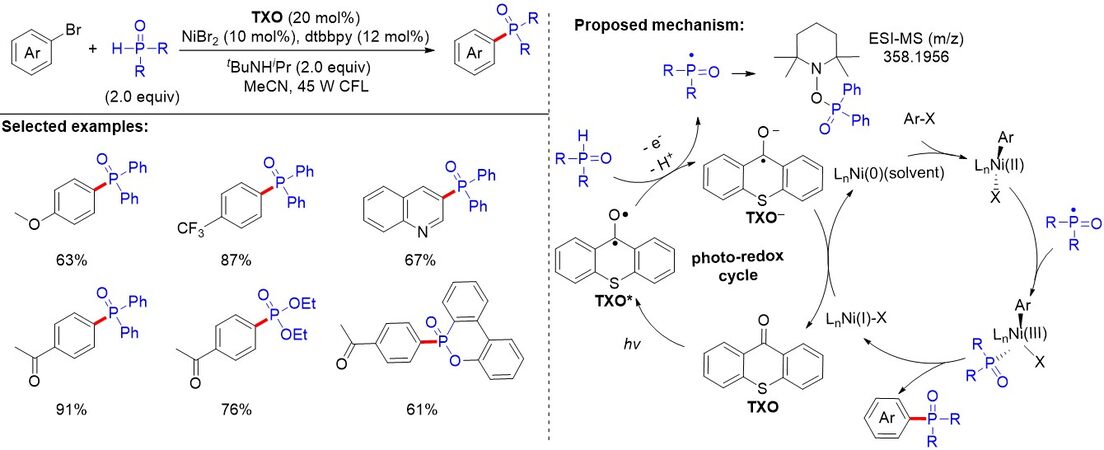
Figure 63. Visible light-induced nickel-catalyzed C(sp2)-P(O) coupling using thioxanthen-9-one as a photoredox catalyst.
Recently, the visible light-driven LMCT-enabled phosphorylation of aryl halides was discovered by
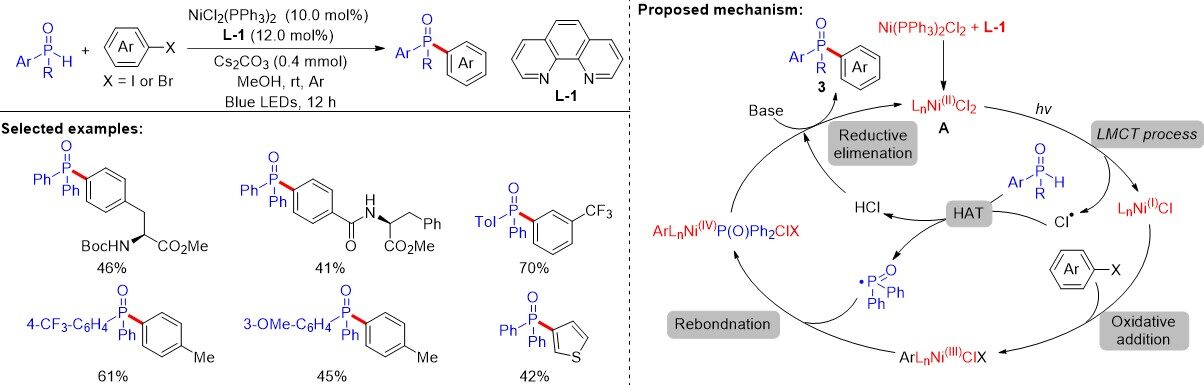
Figure 64. Visible light-induced ligand to metal charge transfer excitation enabled phosphorylation of aryl halides.
Decarboxylative or denitro phosphorylation
Transition metal-catalyzed decarboxylative coupling towards C-C bond construction has been extensively studied. However, comparatively little attention has been focused on C-P bond formation, especially using easily available alkenyl acids as the starting materials. In 2017, Tang et al.[98] reported an efficient protocol for the highly chemo- and stereoselective synthesis of (E)-alkenyl-phosphonates from easily accessible alkenyl acids and H-P(O) compounds using low-cost Cu(OAc)2/K2S2O8 catalytic systems [Figure 65]. Mechanistic studies indicate that acetonitrile is a good solvent and ligand, which could be readily recycled after the reaction. Notably, the cross-coupling of styrenes with H-phosphonates, as well as denitration of β-nitrostyrene with H-phosphonates, is also achieved to give the final products.
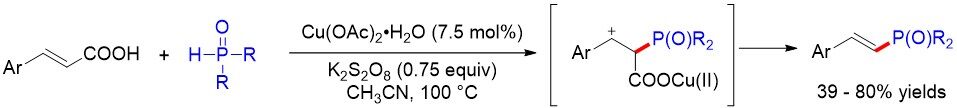
Figure 65. Solvent-controlled copper-catalyzed radical decarboxylative coupling.
β-Nitrostyrenes are useful starting materials in organic synthesis for many classes of compounds. Although the Mn(III)-catalyzed reaction of 2-nitrostyrenes with dialkyl phosphites has been developed by
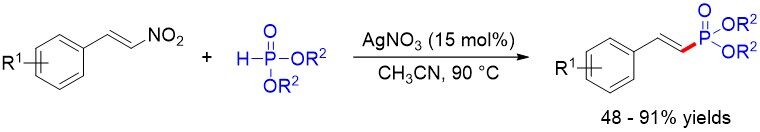
Figure 66. Silver-catalyzed cross-coupling of β-nitrostyrenes with H-phosphites.
Dehydrogenative C-H phosphorylation
Compared to the use of prefunctionalized aromatics, such as aryl halides and boronic acids, as coupling partners, direct C-H functionalization has emerged as an atom-economical and environmentally benign synthetic tool, since it avoids the necessity for the prefunctionalization of the coupling partner. Transition metal-catalyzed C-H bond activation has emerged as an efficient method for the construction of C-P bonds. However, here we mainly discuss P-radical-based C-H dehydrogenative C-H phosphorylation. In recent decades, the development of efficient methods for the introduction of phosphonyl group into those pharmaceutically important molecules has received special attention. Chromenes and coumarins are vital structures in natural products, biological molecules and materials chemistry. In 2012, Zhou et al.[101] reported a Mn(OAc)3-mediated radical phosphorylation at the 3-postion of flavones and coumarins. In 2015, Yuan et al.[102] developed the silver-catalyzed direct C(sp2)-H radical phosphorylation of coumarins with H-phosphites, affording selective coumarin-3-yl phosphonate in moderate to good yields. In 2016, a silver-mediated C-H phosphonylation reaction of ferrocenyl anilides with dialkyl phosphites was developed [Figure 67][103]. The method is free of palladium and copper catalysts and tolerates a variety of functional groups. Mechanistic studies indicate that a radical pathway is involved in these reactions.
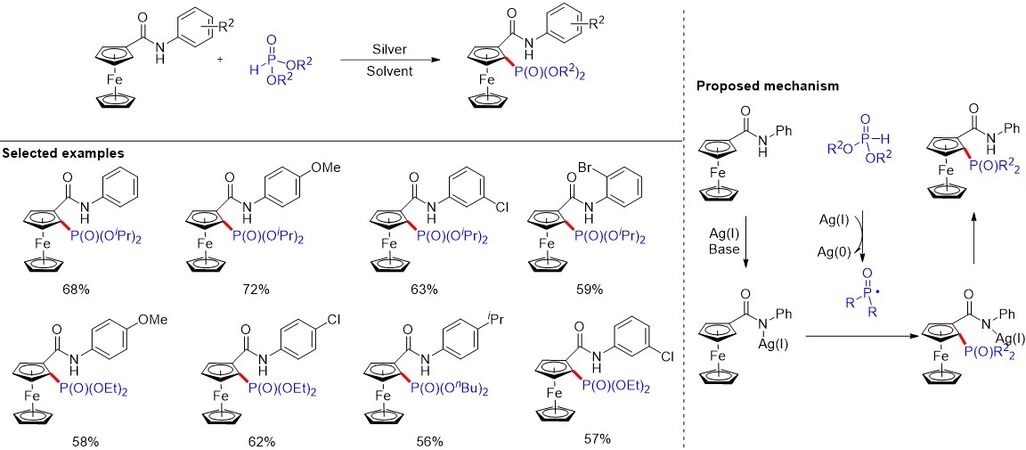
Figure 67. Silver-mediated C-H phosphonylation reaction of ferrocenyl anilides with dialkyl phosphites.
Luo et al.[104] reported the first direct C-H phosphorylation of thiazole derivatives with diarylphosphine oxides without an external oxidant via eosin B catalysis [Figure 68]. The photoredox approach tolerates diverse functional groups and produces a variety of thiazole derivatives bearing electron-donating groups, such as methoxy, methyl and phenyl groups, in medium to excellent yields.
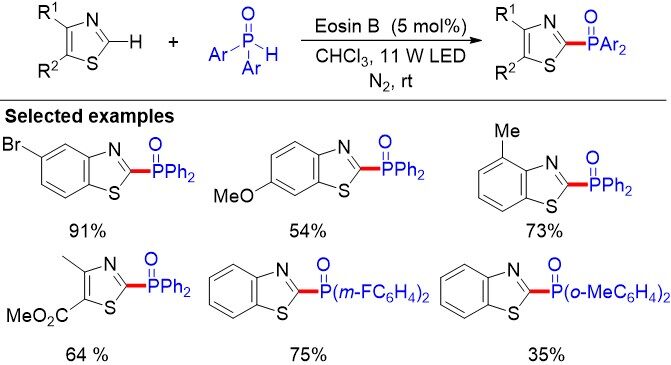
Figure 68. Cross-coupling hydrogen evolution by visible-light photocatalysis toward C(sp2)-P formation.
The C-H phosphonation of the quinoline ring is a highly efficient method for accessing its phosphorylated derivative. The C5-H phosphonation of 8-aminoquinoline amides has been realized by Qiao et al.[105] using silver salts; however, the method suffers from stoichiometric metal salts, a high reaction temperature and a limited substrate scope [Figure 69]. Furthermore, the C4-H phosphonation of 8-aminoquinoline amide remains elusive. Therefore, developing a mild and simple reaction procedure is indispensable for constructing the site-selective C4-H or C5-H phosphonation of 8-aminoquinoline amides. In 2017, the same group described the site-selective C4-H or C5-H phosphonation of 8-aminoquinoline amides via merging photoredox catalysis with transition metal catalysis. In addition, the substrates bearing Me and MeO groups on the quinoline ring could also provide the desired products with good yields. Notably, the strength of the coordination bonds of the bidentate chelated complexes between 8-aminoquinoline and the metal salt determines the regioselectivity at the C4-H or C5-H of 8-aminoquinoline amides. The bidentate chelated complexes with strong Npyr→M bonds give the C4-phophonation product, while those with weak Npyr→M bonds give the C5-phophonation product.
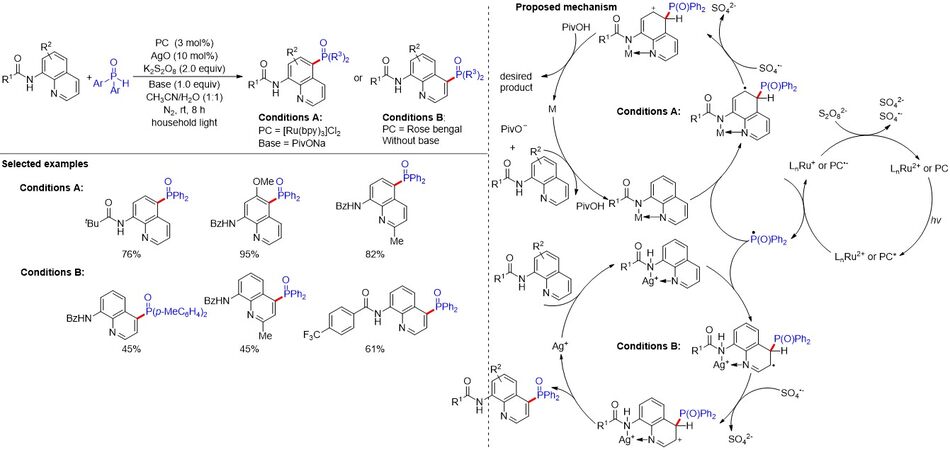
Figure 69. Merging of photoredox and transition metal catalysis: site-selective C4 or C5-H phosphonation of 8-aminoquinoline amides.
According to previous reports, a plausible mechanism is illustrated in Figure 69. Firstly, the excited-state LnRu2+* or PC* was formed under the irradiation of light, which undergoes a reductive quenching process with diarylphosphine oxide to provide the LnRu+ or PC radical anion and an intermediate radical. To complete the photocatalytic cycle, the oxidation of LnRu+ or a PC radical anion by K2S2O8 regenerates the ground-state LnRu2+ or PC. Meanwhile, the addition of the phosphorus radical to the complex of 8-aminoquinoline amide with the transition metal at the C5 position leads to a radical intermediate. After the oxidation by a sulfate radical anion and deprotonation, the product was obtained along with completion of the catalytic cycle of C5 phosphonation. In contrast, the coordination of 8-aminoquinoline amide with Ag(I) affords a chelated intermediate. The addition of the phosphorus radical to the chelated intermediate at the C4 position produces an intermediate radical. Subsequently, oxidation of such an intermediate and following decomposition delivers the desired C4-phophonation product.
Although significant progress has been achieved in the mono-phosphorylation of quinolines, the dephosphorylation of quinolines remains to be addressed due to the importance of organic phosphorus compounds. In 2020, Xiong et al.[106] reported a diphosphonation of quinoline compounds under visible-light irradiation [Figure 70]. This straightforward and environmentally friendly method allows us to access diphosphorylated quinoline compounds. Both electron-donating (-Me or -OMe) and electron-withdrawing groups (-Br or -NO2) at the 6-positions of the quinoline could afford the desired products in moderate to good yields. Unfortunately, some heterocyclic compounds, such as quinoxoline and benzothiophene, could only provide mono-phosphonation products.
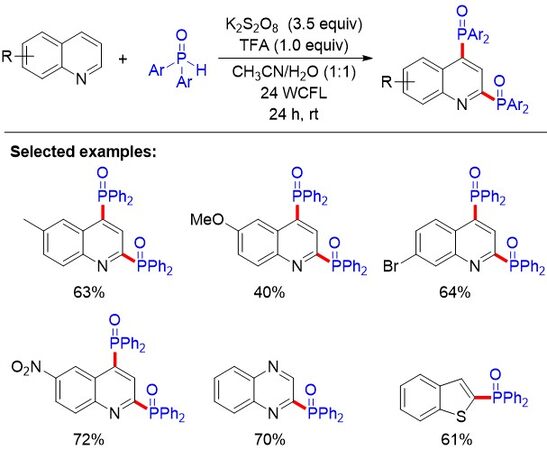
Figure 70. Photocatalytic synthesis of diphosphorous quinoline compounds.
Electrochemical organic synthesis, which uses electrons as clean redox reagents, has been recognized as an efficient and environmentally benign strategy and attracted significant attention in recent years. Although the anodic oxidative phosphonation of arenes as an early example was reported by Effenberger and Kottmann, electrochemical oxidative C-H phosphorylation is still underdeveloped. Recently, as an alternative to traditional transition metal and photoredox catalysis, considerable progress has been achieved regarding electrochemical phosphorylation. In 2019, Li et al.[107] described an efficient method for the intermolecular electrochemically dehydrogenative C-H/P-H cross-coupling of quinoxaline-2(1H)-ones with P(O) compounds in an undivided cell under galvanostatic conditions [Figure 71]. Furthermore, the protocol could be extended to the C(sp3)-H phosphonation of xanthenes. This method features mild conditions, a wide substrate scope and avoids the utilization of transition metal catalysts. Notably, cyclic voltammetry analysis was performed to understand the possible reaction mechanism and a radical pathway for the electrochemically oxidative phosphonation of quinoxaline-2(1H)-one was proposed.
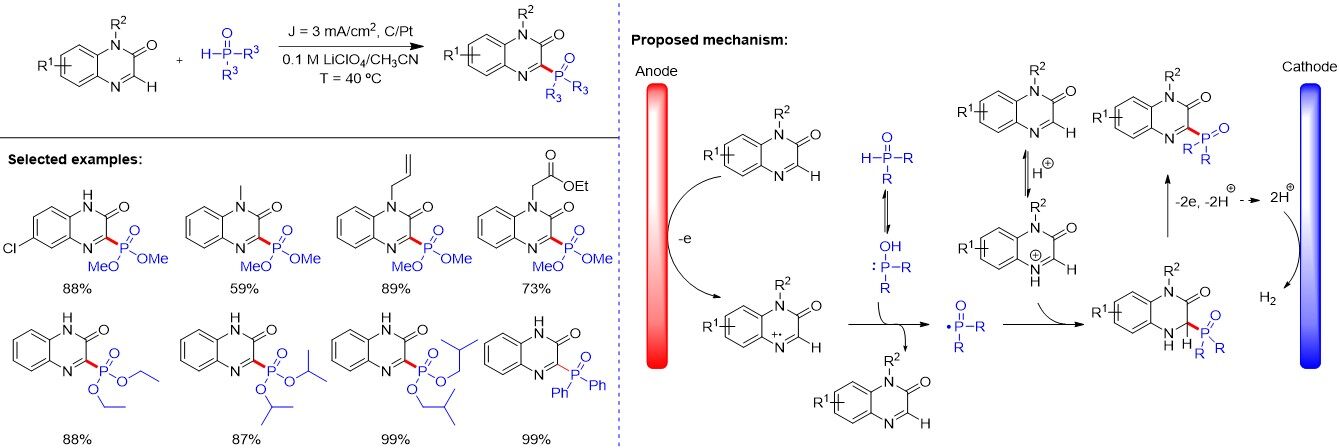
Figure 71. Electrochemically dehydrogenative C-H/P-H cross-coupling.
Similar to the electrochemically oxidative phosphonation of quinoxaline-2(1H)-one, in 2020, Rawat et al.[108] reported the phosphonation of quinoxalines and quinoxalin-2(1H)-ones by employing aerobic oxygen as a green oxidant in the absence of an external base and photocatalyst under visible-light conditions at room temperature [Figure 72]. In addition, alkyl or aryl phosphonates could also be tolerated in this regioselective C-P bond formation reaction under base/ligand-free conditions with good yields. The control experiment, adding radical scavengers TEMPO and BHT to the reaction system under standard conditions, results in no desired products, suggesting the radical pathway. Various phosphonates (iPr, Et, Me, Bu and Bn) reacted with quinoxalines and quinoxalin-2(1H)-ones smoothly and generated the corresponding phosphonated products. A plausible reaction mechanism was proposed [Figure 72]. Initially, the phosphite radical intermediate was derived from alkyl or aryl phosphonate under aerobic conditions in the presence of visible light. The reaction between phosphite radicals and quinoxaline gave a protonated radical cation intermediate in the presence of a hydroperoxy radical. Further HAT and oxidation lead to the formation of the final desired product.
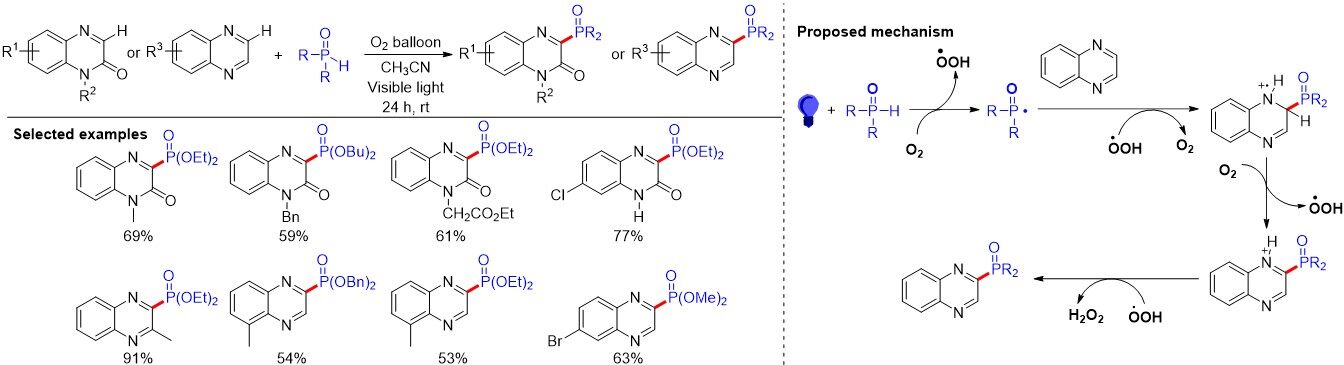
Figure 72. Visible light-induced phosphonation of quinoxalines and quinoxalin-2(1H)-ones under aerobic metal-free conditions.
Indazoles are a class of important N-heterocycles that have received considerable attention due to their numerous pharmaceutical activities. In 2018, the phosphonylation of 2H-indazoles was explored by Singsardar et al.[109] [Figure 73]. The metal-free visible light-induced C-H phosphonylation reaction employed diphenylphosphine oxides as P-radical precursors and Rose Bengal as the organophotoredox catalyst under ambient air at room temperature. A library of diphenyl(2-phenyl-2H-indazol-3-yl)phosphine oxides has been formed with broad functionalities in high yields.
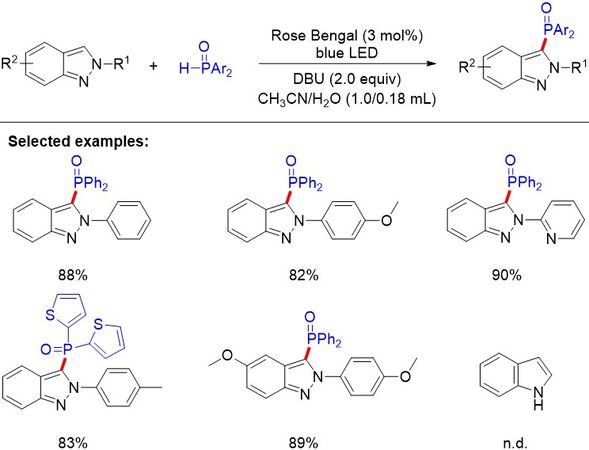
Figure 73. Visible light-induced organophotoredox-catalyzed phosphonylation of 2H-indazoles with diphenylphosphine oxide.
Imidazo[1,2-a]pyridines are ubiquitous and key structural motifs in N-heteroarenes, which are usually found in many commercially available drugs. The direct C-H functionalization of imidazo[1,2-a]pyridines represents an attractive method for the synthesis such highly valuable heteroaromatics. In 2020,
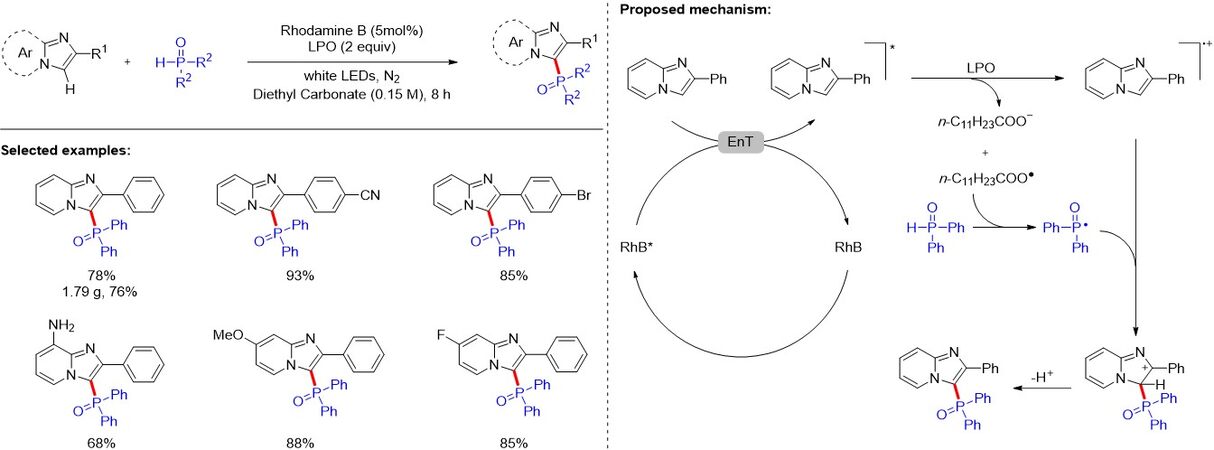
Figure 74. Phosphorylation of imidazo-fused heterocycles under metal-free conditions.
Inspired by their previous work that cobaloxime could activate H-phosphine oxide into radical species under extremely mild reaction conditions, in 2020, Lei et al.[111] finished the phosphorylation of enamines via cobaloxime catalysis under visible-light irradiation [Figure 75]. The oxidation of diphenylphosphine oxide by the excited cobaloxime catalyst produces phosphinoyl radicals via a reductive quenching pathway, which undergoes cross-coupling with various enamines and enamides to give various β-phosphinoyl products in good to excellent yields.
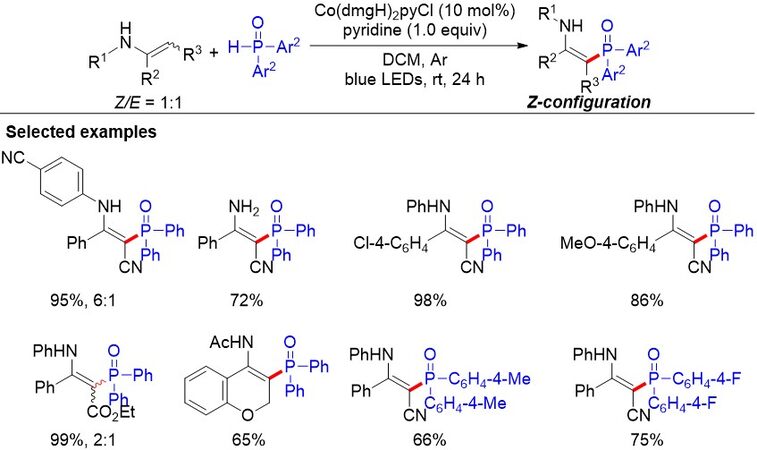
Figure 75. Cobaloxime catalysis for enamine phosphorylation with hydrogen evolution.
PHOSPHORYLATION OF OTHER SUBSTRATES
Carbon-carbon bonds are the most ubiquitous and fundamental chemical bonds in organic molecules. The selective cleavage of C-C bonds and their subsequent transformation remains a major challenge in organic synthesis. In 2017, Li et al.[112] developed a silver-catalyzed oxidative C(sp3)-P bond formation with 1,3-dicarbonyl compounds and H-phosphonates via C-C and P-H bond cleavage [Figure 76]. The protocol exhibits a wide substrate scope, high functional group compatibility and exclusive selectivity. Preliminary mechanistic studies indicate that a radical process and a nucleophilic attack pathway may be involved in the C(sp3)-P bond formation and C(sp3)-C(CO) bond cleavage steps, respectively. A similar work involving Fe-catalyzed and air-mediated C(sp3)-H phosphorylation of 1,3-dicarbonyl compounds involving C-C bond cleavage was reported by Ou et al.[113].
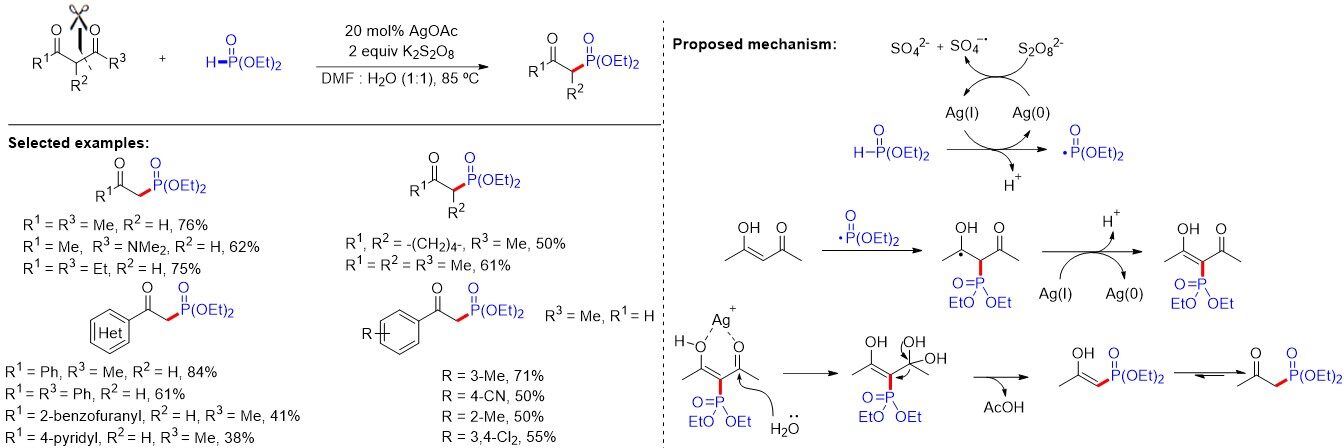
Figure 76. Silver-catalyzed oxidative C(sp3)-P bond formation via C-C and P-H bond cleavages.
In contrast to the above transition metal-catalyzed oxidative C(sp3)-P bond formation via C-C and P-H bond cleavages, in 2019, Zhao et al.[114] reported a photosensitized C(sp3)-C(CO) bond cleavage and C-P bond formation of 1,3-dicarbonyls with arylphosphine oxides [Figure 77]. A series of β-ketophosphine oxides could be obtained in moderate to excellent yields. Control experiments, cyclic voltammetry and Stern-Volmer quenching experiments confirmed that the reaction proceeds via a radical cascade reaction with chemoselective C-C bond cleavage to construct a C-P bond. The SET or the free radical chain reaction affords the critical active intermediates of PhCOO•, β-dicarbonyl and P-radicals with the decomposition of BPO under visible-light irradiation.
Figure 77. Photosensitized C(sp3)-C(CO) bond cleavage and C-P bond formation of 1,3-dicarbonyls with arylphosphine oxides.
A feasible reaction process was proposed. Initially, the excited-state FI* of the photocatalyst FI could be formed under green LED irradiation. Next, there are three main pathways to generate the phosphinoyl radical: (1) to generate the P-radical, the photoexcited FI* promotes the homolytic cleavage of the H-P bond of phosphine oxide via SET [Figure 77]; (2) BPO could be reduced to the pioneer radical PhCOO• by the excited photocatalyst FI* via SET for gaining P-radical; and (3) through a tandem process involving the BPO decomposition and the free radical chain reaction. The active P-radical might also be generated from diphenylphosphine oxide. Simultaneously, β-dicarbonyls react with the FI* to provide the β-dicarbonyl radical. Subsequently, the β-phosphonyl carbon radical intermediate (detected by ESI-MS) and enol could be gained from the reaction between the P-radical with β-diketone or β-dicarbonyl radical. In the presence of BPO or FI•+, the conversion of the β-phosphonyl carbon radical intermediate to the enol intermediate could occur. The electron-deficient carbonyl group was then attacked by H2O to give geminal diols stabilized by the intramolecular hydrogen bonds. The geminal diols underwent a proton transfer and C-C bond cleavage to produce the desired products.
P(O)-O and P(O)-N compounds are significantly important organophosphorus compounds due to their broad applications in pharmaceuticals, agrochemicals, flame retardants and ligand scaffolds. Thus, considerable efforts have been devoted to the development of more efficient and versatile synthetic methodologies to access these compounds. The Atherton-Todd reaction, generating phosphoryl chlorides in situ by the halogenation of HP(O) compounds, is one of the most straightforward methods for the phosphorylation of alcohols and amines. However, the use of highly toxic CCl4 as the halogenating reagent and the solvent make the applications of this reaction rather limited. In 2020, Ou et al.[115] reported a phosphoryl radical-initiated Atherton-Todd-type reaction under air [Figure 78]. Notably, the utilization of less toxic CHCl3 as the halogenating reagent instead of CCl4 and air as the radical initiator enables the method to be more environmentally and user friendly. The inexpensive, simple and efficient synthesis of phosphinates, phosphinic amides and phosphoramidates with high functional group tolerance exhibits the potential application of this approach in organic chemistry. A plausible mechanism is illustrated in Figure 78. Initially, a phosphoryl radical is generated by the auto-oxidation of the phosphoryl anion with O2, which is generated from the deprotonation of the P(O)-H compound. The chlorine abstraction of CHCl3 by the phosphoryl radical then gives rise to the dichloromethyl radical and phosphoryl chloride. Eventually, a nucleophilic substitution reaction of a nucleophile reagent, such as alcohols and amines, with phosphoryl chloride affords the desired product.
Figure 78. A phosphoryl radical-initiated Atherton-Todd-type reaction under open air.
Thiophosphates are also one of the most important classes of phosphorus-containing scaffolds and frequently occur in pharmaceutically intriguing molecules. Therefore, the development of practical, efficient and green protocols for the synthesis of thiophosphates constitutes an important goal in contemporary organic chemistry. In 2018, Zhang et al.[116] reported a visible-light photoredox-catalyzed thiophosphate synthesis with methylene blue, leading to the products in moderate to good yields [Figure 79]. Moreover, Stern-Volmer fluorescence quenching experiments were conducted to illustrate the possible reaction mechanism.
Figure 79. Visible-light photoredox-catalyzed synthesis of thiophosphonate using methylene blue.
Phosphonated azacyclic compounds containing P(O)-N bonds have shown excellent applications in materials chemistry as organic semiconductors and organic light-emitting diodes due to their excellent electron donor-acceptor properties. Therefore, the construction of P(O)-N bonds is synthetically important. Although some pioneering work has been established on the electrosynthesis of P(O)-N bonds by the direct oxidative cross coupling of N-H/P-H, the substrates were only restricted to alkyl primary and secondary amines. In 2020, Dong et al.[117] presented an electrochemical dehydrogenative phosphorylation of nitrogen-containing heterocycles with an imidazolium-based ionic liquid (1,3-dimethylimidazolium iodide) as an electrolyte under mild reaction conditions [Figure 80]. Diverse phosphorylation products of substituted carbazoles and indoles are constructed in moderate to excellent yields. Cyclic voltammogram experiments were also performed to investigate the reaction mechanism and a plausible mechanism was proposed for the reaction. Initially, the iodide radical is generated by the oxidation of the iodide anion at the anode. The P(O)-H compound then interacts with an iodide radical, leading to the phosphonyl radical, which further couples with an iodide radical to form the intermediate P(O)-I compound. Meanwhile, the N-heterocycle anion is generated by the deprotonation of the starting material P(O)-H compound with Cs2CO3. The nucleophilic attack of P(O)-I compound by the N-heterocycle anion results in the final desired product. Notably, reduction of the proton at the cathode furnishes hydrogen gas.
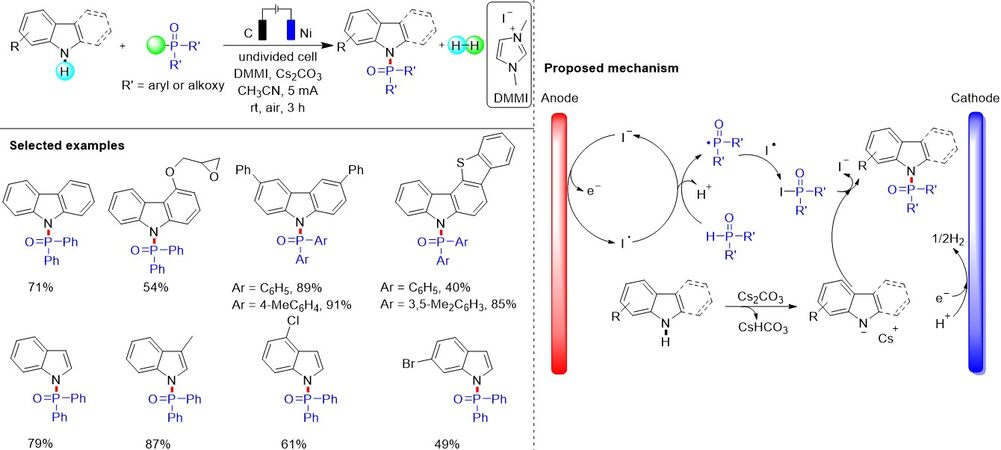
Figure 80. Electrochemical oxidative dehydrogenative phosphorylation of N-heterocycles.
CONCLUSIONS AND OUTLOOK
The growing awareness of the importance of organophosphorus compounds gives rise to a strong and renewed relevance to the development of P-centered radical-based reactions. Owing to the versatile reactivity of P-centered radicals in various transformations, considerable progress has been achieved in transition metal-mediated and photoredox-catalyzed phosphorylation reactions. In addition, it is noteworthy that electrochemical phosphorylation has also emerged as an efficient approach for the construction of organophosphorus compounds. In particular, over the past decade, the phosphorus radical-initiated phosphorylation of unsaturated compounds, such as alkenes, alkynes and isocyanides, through merging with transition metal catalysis, photocatalysis and electrochemistry, has become one of the most appealing strategies to build structurally diverse organophosphorus compounds. Furthermore, cross-coupling reactions with X-H (X=C, N, O or S) bonds and (pseudo)halides, involving phosphorus radicals, have also transformed into a powerful protocol for the efficient synthesis of otherwise difficult to access organophosphorus compounds.
Despite the myriad of recent advances that have been made in this fast-growing research area, several limitations, restrictions and challenges remain to be addressed in the future in order to improve the practicality and versatility of these methods. First of all, the types and sources of phosphorus radicals are still limited, thus expanding the types and sources of phosphorus radical is worth exploring. Second, the usage of transition metal salts or organometallic reagents in most phosphorylation reactions, which might hamper their further application in industry, makes the development of more environmentally sustainable approaches essential. Third, the development of enantioselective C-P bond formation reactions is another exciting field. Overall, there are still many challenges and problems in the construction of organophosphorus compounds that need to be resolved and deserve the attention of chemists.
DECLARATIONS
AcknowledgmentsWe sincerely thank all leading chemists and co-workers involved in the development of phosphorus radical-based phosphorylation reactions.
Authors’ contributionsWrote the paper: Liu J, Xiao HZ, Fu Q
Guided this work, gave valuable suggestions and discussion for the review and revised the paper: Fu Q,
Not applicable.
Financial support and sponsorshipThis work was supported by Natural Science Foundation of China (No. 21772129; No. 22001220), and the Fundamental Research Funds for the Central Universities.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Tang W, Zhang X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem Rev 2003;103:3029-70.
2. Demmer CS, Krogsgaard-Larsen N, Bunch L. Review on modern advances of chemical methods for the introduction of a phosphonic acid group. Chem Rev 2011;111:7981-8006.
3. Duffy MP, Delaunay W, Bouit PA, Hissler M. π-Conjugated phospholes and their incorporation into devices: components with a great deal of potential. Chem Soc Rev 2016;45:5296-310.
4. der Jeught S, Stevens CV. Direct phosphonylation of aromatic azaheterocycles. Chem Rev 2009;109:2672-702.
5. Shao C, Xu W, Li L, Zhang X. Recent advances of transition metal-catalyzed P-C coupling reactions. Chin J Org Chem 2017;37:335.
6. Gao Y, Tang G, Zhao Y. Recent progress toward organophosphorus compounds based on phosphorus-centered radical difunctionalizations. Phosphorus, Sulfur, and Silicon and the Related Elements 2017;192:589-96.
7. Lan X, Wang N, Xing Y. Recent Advances in Radical Difunctionalization of Simple Alkenes: Recent Advances in Radical Difunctionalization of Simple Alkenes. Eur J Org Chem 2017;2017:5821-51.
8. Gao Y, Tang G, Zhao Y. Recent advances of phosphorus-centered radical promoted difunctionalization of unsaturated carbon-carbon bonds. Chin J Org Chem 2018;38:62.
9. Cai B, Xuan J, Xiao W. Visible light-mediated C P bond formation reactions. Science Bulletin 2019;64:337-50.
10. Chen L, Liu X, Zou Y. Recent advances in the construction of phosphorus-substituted heterocycles, 2009-2019. Adv Synth Catal 2020;362:1724-818.
11. Ung SP, Mechrouk VA, Li C. Shining light on the light-bearing element: a brief review of photomediated C-H phosphorylation reactions. Synthesis 2021;53:1003-22.
12. Leca D, Fensterbank L, Lacôte E, Malacria M. Recent advances in the use of phosphorus-centered radicals in organic chemistry. Chem Soc Rev 2005;34:858-65.
13. Rueping M, Zhu S, Koenigs RM. Photoredox catalyzed C-P bond forming reactions-visible light mediated oxidative phosphonylations of amines. Chem Commun (Camb) 2011;47:8679-81.
14. Kingston C, Palkowitz MD, Takahira Y, et al. A survival guide for the "Electro-curious". Acc Chem Res 2020;53:72-83.
15. Budnikova YH, Gryaznova TV, Grinenko VV, Dudkina YB, Khrizanforov MN. Eco-efficient electrocatalytic C-P bond formation. Pure and Applied Chemistry 2017;89:311-30.
16. Bezzenine-Lafollée S, Gil R, Prim D, Hannedouche J. First-row late transition metals for catalytic alkene hydrofunctionalisation: recent advances in C-N, C-O and C-P bond formation. Molecules 2017;22:1901.
17. Yoo W, Kobayashi S. Hydrophosphinylation of unactivated alkenes with secondary phosphine oxides under visible-light photocatalysis. Green Chem 2013;15:1844.
18. Wang N, Ye L, Li Z, et al. Hydrofunctionalization of alkenols triggered by the addition of diverse radicals to unactivated alkenes and subsequent remote hydrogen atom translocation. Org Chem Front 2018;5:2810-4.
19. Yuan Z, Wang S, Li M, et al. Visible light induced hydrophosphinylation of unactivated alkenes catalyzed by salicylaldehyde. Green Chem 2021;23:3600-6.
20. Li YM, Sun M, Wang HL, Tian QP, Yang SD. Direct annulations toward phosphorylated oxindoles: silver-catalyzed carbon-phosphorus functionalization of alkenes. Angew Chem Int Ed Engl 2013;52:3972-6.
21. Li Y, Shen Y, Chang K, Yang S. Metal-free oxidative arylphosphination of activated N-substituted-N-arylacrylamide derivatives using K2S2O8. Tetrahedron 2014;70:1991-6.
22. Zhao J, Li P, Li X, Xia C, Li F. Straightforward synthesis of functionalized chroman-4-ones through cascade radical cyclization-coupling of 2-(allyloxy)arylaldehydes. Chem Commun (Camb) 2016;52:3661-4.
23. Liu Q, Lu W, Xie G, Wang X. Metal-free synthesis of phosphinoylchroman-4-ones via a radical phosphinoylation-cyclization cascade mediated by K2S2O8. Beilstein J Org Chem 2020;16:1974-82.
24. Zhang HY, Mao LL, Yang B, Yang SD. Copper-catalyzed radical cascade cyclization for the synthesis of phosphorated indolines. Chem Commun (Camb) 2015;51:4101-4.
25. Liang D, Ge D, Lv Y, Huang W, Wang B, Li W. Silver-catalyzed radical arylphosphorylation of unactivated alkenes: synthesis of 3-phosphonoalkyl indolines. J Org Chem 2018;83:4681-91.
26. Liu X, Sun K, Lv Q, et al. Silver-mediated radical phosphorylation/cyclization of N -allylbenzamides to access phosphoryl-substituted dihydroisoquinolones. New J Chem 2019;43:12221-4.
27. Liu X, Sun K, Chen X, et al. Visible-light-promoted transition-metal-free approach toward phosphoryl-substituted dihydroisoquinolones via cascade phosphorylation/cyclization of N-allylbenzamides. Adv Synth Catal 2019;361:3712-7.
28. Liu J, Zhao S, Song W, et al. Silver-catalyzed carbon-phosphorus functionalization for polyheterocycle formation. Adv Synth Catal 2017;359:609-15.
29. Gorre R, Enagandhula D, Balasubramanian S, Akondi SM. Visible-light-driven metal-free aerobic synthesis of highly diastereoselective phosphinoylpyrroloindoles. Org Biomol Chem 2020;18:1354-8.
30. Li Y, Zhu Y, Yang S. Visible-light-induced tandem phosphorylation cyclization of vinyl azides under mild conditions. Org Chem Front 2018;5:822-6.
31. Sun K, Si Y, Chen X, et al. Synthesis of phosphoryl-substituted benzimidazo[2,1-a]isoquinolin-6(5 H)-ones from 2-arylbenzoimidazoles and diarylphosphine oxides. Asian J Org Chem 2019;8:2042-5.
32. Jiang SS, Xiao YT, Wu YC, Luo SZ, Song RJ, Li JH. Manganese(III)-promoted tandem phosphinoylation/cyclization of 2-arylindoles/2-arylbenzimidazoles with disubstituted phosphine oxides. Org Biomol Chem 2020;18:4843-7.
33. Fu Q, Bo ZY, Ye JH, et al. Transition metal-free phosphonocarboxylation of alkenes with carbon dioxide via visible-light photoredox catalysis. Nat Commun 2019;10:3592.
34. Jiang Y, Liu J, Fu Q, Yu Y, Yu D. Visible-light-driven phosphonoalkylation of alkenes. Synlett 2021;32:378-82.
35. Chu XQ, Zi Y, Meng H, Xu XP, Ji SJ. Radical phosphinylation of α,α-diaryl allylic alcohols with concomitant 1,2-aryl migration. Chem Commun (Camb) 2014;50:7642-5.
36. Mi X, Wang C, Huang M, Wu Y, Wu Y. Silver-catalyzed carbonphosphonation of α,α-diaryl allylic alcohols: synthesis of β-aryl-γ-ketophosphonates. Org Biomol Chem 2014;12:8394-7.
37. Yin Y, Weng WZ, Sun JG, Zhang B. Eosin Y-catalyzed, visible-light-promoted carbophosphinylation of allylic alcohols via a radical neophyl rearrangement. Org Biomol Chem 2018;16:2356-61.
38. Li C, Qi Z, Yang Q, Qiang X, Yang S. Visible-light-catalyzed phosphonation-annulation: an efficient strategy to synthesize β-Phosphonopyrrolidines and β-phosphonolactones: visible-light-catalyzed phosphonation-annulation: an efficient strategy to synthesize β-phosphonopyrrolidines and β-phosphonolactones†. Chin J Chem 2018;36:1052-8.
39. Wang C, Huang X, Liu X, Gao S, Zhao B, Yang S. Photo-induced phosphorus radical involved semipinacol rearrangement reaction: highly synthesis of γ-oxo-phosphonates. Chinese Chemical Letters 2020;31:677-80.
40. Jin S, Sun S, Yu JT, Cheng J. The silver-promoted phosphonation/alkynylation of alkene proceeding with radical 1,2-alkynyl migration. J Org Chem 2019;84:11177-85.
41. Zhang PZ, Zhang L, Li JA, Shoberu A, Zou JP, Zhang W. Phosphinoyl radical initiated vicinal cyanophosphinoylation of alkenes. Org Lett 2017;19:5537-40.
42. Zhang G, Fu L, Chen P, Zou J, Liu G. Proton-coupled electron transfer enables tandem radical relay for asymmetric copper-catalyzed phosphinoylcyanation of styrenes. Org Lett 2019;21:5015-20.
43. He Y, Won J, Kim J, et al. One-pot bifunctionalization of unactivated alkenes, P(O)-H compounds, and N-methoxypyridinium salts for the construction of β-pyridyl alkylphosphonates. Org Chem Front 2018;5:2595-603.
44. Buquoi JQ, Lear JM, Gu X, Nagib DA. Heteroarene phosphinylalkylation via a catalytic, polarity-reversing radical cascade. ACS Catal 2019;9:5330-5.
45. Lear JM, Buquoi JQ, Gu X, Pan K, Mustafa DN, Nagib DA. Multi-component heteroarene couplings via polarity-reversed radical cascades. Chem Commun (Camb) 2019;55:8820-3.
46. Shen J, Zhang Y, Yu Y, Wang M. Metal-free visible-light-induced photoredox-catalyzed intermolecular pyridylation/phosphinoylation of alkenes. Org Chem Front 2021;8:901-7.
47. Maryanoff BE, Reitz AB. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem Rev 1989;89:863-927.
48. Wei W, Ji JX. Catalytic and direct oxyphosphorylation of alkenes with dioxygen and H-phosphonates leading to β-ketophosphonates. Angew Chem Int Ed Engl 2011;50:9097-9.
49. Zhou M, Zhou Y, Song Q. Cu/Fe-cocatalyzed formation of β-Ketophosphonates by a domino knoevenagel-decarboxylation-oxyphosphorylation sequence from aromatic aldehydes and H-phosphonates. Chemistry 2015;21:10654-9.
50. Chen X, Chen X, Li X, et al. Acetonitrile-dependent oxyphosphorylation: a mild one-pot synthesis of β-ketophosphonates from alkenyl acids or alkenes. Tetrahedron 2017;73:2439-46.
51. Fu Q, Yi D, Zhang Z, et al. Copper-catalyzed aerobic oxidative coupling of ketones with P(O)-H compounds leading to β-ketophosphine oxides. Org Chem Front 2017;4:1385-9.
52. Zhang Z, Yi D, Fu Q, et al. Copper catalyzed one-pot synthesis of β-ketophosphine oxides from ketones and H-phosphine oxides. Tetrahedron Letters 2017;58:2417-20.
53. Liang W, Zhang Z, Yi D, et al. Copper-catalyzed direct oxyphosphorylation of enamides with P(O)-H compounds and dioxygen. Chin J Chem 2017;35:1378-82.
54. Nan G, Yue H. Direct synthesis of β-ketophosphine oxides via copper-catalyzed difunctionalization of alkenes with H-phosphine oxides and dioxygen. Tetrahedron Letters 2018;59:2071-4.
55. Wang H, Fu Q, Zhang Z, Gao M, Ji J, Yi D. Hydrochloric acid-promoted copper/iron-cocatalyzed deesterifica-tive oxyphosphorylation of 2-substituted acrylates with H-phosphine oxides. Chin J Org Chem 2018;38:1977.
56. Feng S, Li J, He F, et al. A copper-catalyzed radical coupling/fragmentation reaction: efficient access to β-oxophosphine oxides. Org Chem Front 2019;6:946-51.
57. Gu J, Cai C. Cu(I)/Fe(III)-Catalyzed C-P cross-coupling of styrenes with H-phosphine oxides: a facile and selective synthesis of alkenylphosphine oxides and β-ketophosphonates. Org Biomol Chem 2017;15:4226-30.
58. Moghaddam FM, Daneshfar M, Azaryan R, Pirat J. Copper ferrite nanoparticles catalyzed formation of β-Ketophosphonates via oxyphosphorylation of styrenes with H-phosphonates: a DFT study on UV-vis absorption spectra. Catalysis Communications 2020;141:106015.
59. Shi Y, Chen R, Guo K, et al. Visible light-promoted metal-free aerobic oxyphosphorylation of olefins: a facile approach to β-ketophosphine oxides. Tetrahedron Letters 2018;59:2062-5.
60. Qian HF, Li CK, Zhou ZH, Tao ZK, Shoberu A, Zou JP. Visible light-mediated photocatalytic metal-free cross-coupling reaction of alkenyl carboxylic acids with diarylphosphine oxides leading to β-ketophosphine oxides. Org Lett 2018;20:5947-51.
61. Jung HI, Kim DY. Visible light-mediated photocatalytic phosphorylation of vinyl azides: A mild synthesis of β-ketophosphine oxides. Synthetic Communications 2020;50:380-7.
62. Li M, Zhang Q, Hu D, et al. Catalyst-free direct difunctionalization of alkenes with H-phosphine oxides and dioxygen: a facile and green approach to β-hydroxyphosphine oxides. Tetrahedron Letters 2016;57:2642-6.
63. Peng P, Lu Q, Peng L, Liu C, Wang G, Lei A. Dioxygen-induced oxidative activation of a P-H bond: radical oxyphosphorylation of alkenes and alkynes toward β-oxy phosphonates. Chem Commun (Camb) 2016;52:12338-41.
64. Yi D, Fu Q, Chen S, et al. Copper-catalyzed direct hydroxyphosphorylation of electron-deficient alkenes with H-phosphine oxides and dioxygen. Tetrahedron Letters 2017;58:2058-61.
65. Chen Y, Chen Y, Lu S, Li Z. Copper-catalyzed three-component phosphorylation-peroxidation of alkenes. Org Chem Front 2018;5:972-6.
66. Shen J, Xiao B, Hou Y, et al. Cobalt(II)-catalyzed bisfunctionalization of alkenes with diarylphosphine oxide and peroxide. Adv Synth Catal 2019;361:5198-209.
67. Zhou SF, Li DP, Liu K, Zou JP, Asekun OT. Direct radical acetoxyphosphorylation of styrenes mediated by manganese(III). J Org Chem 2015;80:1214-20.
68. Ryzhakov D, Jarret M, Baltaze JP, Guillot R, Kouklovsky C, Vincent G. Synthesis of 3,3-spirocyclic 2-phosphonoindolines via a dearomative addition of phosphonyl radicals to indoles. Org Lett 2019;21:4986-90.
69. Yang B, Hou S, Ding S, et al. Cerium(IV)-promoted phosphinoylation-nitratation of alkenes. Adv Synth Catal 2018;360:4470-4.
70. Zhang C, Li Z, Zhu L, Yu L, Wang Z, Li C. Silver-catalyzed radical phosphonofluorination of unactivated alkenes. J Am Chem Soc 2013;135:14082-5.
71. Li JA, Zhang PZ, Liu K, Shoberu A, Zou JP, Zhang W. Phosphinoyl radical-initiated α,β-aminophosphinoylation of alkenes. Org Lett 2017;19:4704-6.
72. Wang Y, Wang W, Tang R, Liu Z, Tao W, Fang Z. Iron(III)-catalyzed radical α,β-aminophosphinoylation of styrenes with diphenylphosphine oxides and anilines. Org Biomol Chem 2018;16:7782-6.
73. Xu J, Li X, Gao Y, et al. Mn(III)-mediated phosphonation-azidation of alkenes: a facile synthesis of β-azidophosphonates. Chem Commun (Camb) 2015;51:11240-3.
74. Ma X, Chiou M, Ge L, et al. Iron phthalocyanine-catalyzed radical phosphinoylazidation of alkenes: a facile synthesis of β-azido-phosphine oxide with a fast azido transfer step. Chinese Journal of Catalysis 2021;42:1634-40.
75. Tao ZK, Li CK, Zhang PZ, Shoberu A, Zou JP, Zhang W. Phosphinoyl radical-Initiated 1,2-bifunctional thiocyanodiphenylphosphinoylation of alkenes. J Org Chem 2018;83:2418-24.
76. Huang T, Saga Y, Guo H, Yoshimura A, Ogawa A, Han LB. Radical hydrophosphorylation of alkynes with R2P(O)H generating alkenylphosphine oxides: scope and limitations. J Org Chem 2018;83:8743-9.
77. Wang H, Li Y, Tang Z, et al. Z-Selective addition of diaryl phosphine oxides to alkynes via photoredox catalysis. ACS Catal 2018;8:10599-605.
78. Liu WQ, Lei T, Zhou S, et al. Cobaloxime catalysis: selective synthesis of alkenylphosphine oxides under visible light. J Am Chem Soc 2019;141:13941-7.
79. Hou H, Zhou B, Wang J, et al. Stereo- and Regioselective cis-hydrophosphorylation of 1,3-enynes enabled by the visible-light irradiation of NiCl2(PPh3)2. Org Lett 2021;23:2981-7.
80. Zhang P, Zhang L, Gao Y, et al. Copper-catalyzed tandem phosphination-decarboxylation-oxidation of alkynyl acids with H-phosphine oxides: a facile synthesis of β-ketophosphine oxides. Chem Commun (Camb) 2015;51:7839-42.
81. Chen X, Li X, Chen XL, et al. A one-pot strategy to synthesize β-ketophosphonates: silver/copper catalyzed direct oxyphosphorylation of alkynes with H-phosphonates and oxygen in the air. Chem Commun (Camb) 2015;51:3846-9.
82. Yi N, Wang R, Zou H, He W, Fu W, He W. Copper/Iron-catalyzed aerobic oxyphosphorylation of terminal alkynes leading to β-ketophosphonates. J Org Chem 2015;80:5023-9.
83. Zhou M, Chen M, Zhou Y, et al. β-ketophosphonate formation via aerobic oxyphosphorylation of alkynes or alkynyl carboxylic acids with H-phosphonates. Org Lett 2015;17:1786-9.
84. Zhong W, Tan T, Shi L, Zeng X. Base-promoted direct oxyphosphorylation of alkynes with h-phosphine oxides in the presence of water. Synlett 2018;29:1379-84.
85. Tao ZK, Li CK, Li JA, Shoberu A, Zhang W, Zou JP. Copper-catalyzed vicinal cyano-, thiocyano-, and chlorophosphorylation of alkynes: a phosphinoyl radical-initiated approach for difunctionalized alkenes. Org Lett 2021;23:4342-7.
86. Zhang P, Gao Y, Zhang L, et al. Copper-Catalyzed Cycloaddition between secondary phosphine oxides and alkynes: synthesis of benzophosphole oxides. Adv Synth Catal 2016;358:138-42.
87. Quint V, Morlet-Savary F, Lohier JF, Lalevée J, Gaumont AC, Lakhdar S. Metal-Free, Visible light-photocatalyzed synthesis of benzo[B]phosphole oxides: synthetic and mechanistic investigations. J Am Chem Soc 2016;138:7436-41.
88. Zhu XT, Zhao Q, Liu F, et al. Silver-mediated radical 5-exo-dig cyclization of 2-alkynylbenzonitriles: synthesis of phosphinylated 1-indenones. Chem Commun (Camb) 2017;53:6828-31.
89. Hou H, Xu Y, Yang H, et al. Visible-light mediated hydrosilylative and hydrophosphorylative cyclizations of enynes and dienes. Org Lett 2020;22:1748-53.
90. Huang WB, Ren FY, Wang MW, Qiu LQ, Chen KH, He LN. Cu(II)-catalyzed phosphonocarboxylative cyclization reaction of propargylic amines and phosphine oxide with CO2. J Org Chem 2020;85:14109-20.
91. Liu XC, Chen XL, Liu Y, et al. Visible-light-induced metal-free synthesis of 2-phosphorylated thioflavones in water. ChemSusChem 2020;13:298-303.
92. Li CX, Tu DS, Yao R, Yan H, Lu CS. Visible-light-induced cascade reaction of isocyanides and N-arylacrylamides with diphenylphosphine oxide via radical C-P and C-C bond formation. Org Lett 2016;18:4928-31.
93. Yang W, Li B, Zhang M, et al. Metal-free photo-induced radical C-P and C-S bond formation for the synthesis of 2-phosphoryl benzothiazoles. Chinese Chemical Letters 2020;31:1313-6.
95. Liao LL, Gui YY, Zhang XB, et al. Phosphorylation of alkenyl and aryl C-O bonds via photoredox/nickel dual catalysis. Org Lett 2017;19:3735-8.
96. Zhu DL, Jiang S, Wu Q, et al. Visible-light-induced nickel-catalyzed P(O)-C(sp2) coupling using thioxanthen-9-one as a photoredox catalysis. Org Lett 2021;23:160-5.
97. Hou H, Zhou B, Wang J, et al. Visible-light-induced ligand to metal charge transfer excitation enabled phosphorylation of aryl halides. Chem Commun (Camb) 2021;57:5702-5.
98. Tang L, Wen L, Sun T, et al. Solvent-controlled copper-catalyzed radical decarboxylative coupling for alkenyl C(sp 2 )-P bond formation. Asian J Org Chem 2017;6:1683-92.
99. Xue JF, Zhou SF, Liu YY, Pan X, Zou JP, Asekun OT. Manganese(III)-mediated alkenyl C(sp2)-P bond formation from the reaction of β-nitrostyrenes with dialkyl phosphites. Org Biomol Chem 2015;13:4896-902.
100. Yuan J, Yang L, Mao P, Qu L. Silver-catalyzed synthesis of 2-arylvinylphosphonates by cross-coupling of β-nitrostyrenes with H-phosphites. RSC Adv 2016;6:87058-65.
101. Zhou P, Jiang Y, Zou J, Zhang W. Manganese(III) acetate mediated free-radical phosphonylation of flavones and coumarins. Synthesis 2012;44:1043-50.
102. Yuan J, Li Y, Yang L, et al. Silver-catalyzed direct Csp2-H radical phosphorylation of coumarins with H-phosphites. Tetrahedron 2015;71:8178-86.
103. Yu X, Ge B, Zhang Y, Wang X, Wang D. Silver-Mediated Phosphonylation of C(sp2)-H Bonds with P-H bonds: Direct C-H functionalization of ferrocenyl anilides and dialkyl phosphites under palladium- and copper-free conditions. Asian J Org Chem 2016;5:1253-9.
104. Luo K, Chen YZ, Yang WC, Zhu J, Wu L. Cross-coupling hydrogen evolution by visible light photocatalysis toward C(sp(2))-P FOrmation: metal-free C-H functionalization of thiazole derivatives with diarylphosphine oxides. Org Lett 2016;18:452-5.
105. Qiao H, Sun S, Zhang Y, et al. Merging photoredox catalysis with transition metal catalysis: site-selective C4 or C5-H phosphonation of 8-aminoquinoline amides. Org Chem Front 2017;4:1981-6.
106. Xiong Y, Zhang Y, Qi L, Jiang M, Zhang J, Wang T. Photocatalytic synthesis of diphosphorous quinoline compounds. Asian J Org Chem 2020;9:292-5.
107. Li K, Jiang Y, Xu K, Zeng C, Sun B. Electrochemically dehydrogenative C-H/P-H cross-coupling: effective synthesis of phosphonated quinoxalin-2(1 H)-ones and xanthenes. Green Chem 2019;21:4412-21.
108. Rawat D, Kumar R, Subbarayappa A. Visible-light induced phosphonation of quinoxalines and quinoxalin-2(1 H)-ones under aerobic metal-free conditions. Green Chem 2020;22:6170-5.
109. Singsardar M, Dey A, Sarkar R, Hajra A. Visible-light-induced organophotoredox-catalyzed phosphonylation of 2 H-indazoles with diphenylphosphine oxide. J Org Chem 2018;83:12694-701.
110. Gao F, Sun K, Chen XL, et al. Visible-light-induced phosphorylation of imidazo-fused heterocycles under metal-free conditions. J Org Chem 2020;85:14744-52.
111. Lei T, Liang G, Cheng YY, Chen B, Tung CH, Wu LZ. Cobaloxime catalysis for enamine phosphorylation with hydrogen evolution. Org Lett 2020;22:5385-9.
112. Li L, Huang W, Chen L, Dong J, Ma X, Peng Y. Silver-catalyzed oxidative C(sp3)-P bond formation through C-C and P-H bond cleavage. Angew Chem Int Ed Engl 2017;56:10539-44.
113. Ou Y, Huang Y, Liu Y, et al. Iron-Catalyzed and Air-Mediated C(sp3)-H phosphorylation of 1,3-dicarbonyl compounds involving C-C bond cleavage. Adv Synth Catal 2020;362:5783-7.
114. Zhao X, Huang M, Li Y, Zhang J, Kim JK, Wu Y. Stepwise photosensitized C(sp3)-C(CO) bond cleavage and C-P bond formation of 1,3-dicarbonyls with arylphosphine oxides. Org Chem Front 2019;6:1433-7.
115. Ou Y, Huang Y, He Z, et al. A phosphoryl radical-initiated Atherton-Todd-type reaction under open air. Chem Commun (Camb) 2020;56:1357-60.
116. Zhang H, Zhan Z, Lin Y, et al. Visible light photoredox catalyzed thiophosphate synthesis using methylene blue as a promoter. Org Chem Front 2018;5:1416-22.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu J, Xiao HZ, Fu Q, Yu DG. Advances in radical phosphorylation from 2016 to 2021. Chem Synth 2021;1:9. http://dx.doi.org/10.20517/cs.2021.07
AMA Style
Liu J, Xiao HZ, Fu Q, Yu DG. Advances in radical phosphorylation from 2016 to 2021. Chemical Synthesis. 2021; 1(1): 9. http://dx.doi.org/10.20517/cs.2021.07
Chicago/Turabian Style
Liu, Jie, Han-Zhi Xiao, Qiang Fu, Da-Gang Yu. 2021. "Advances in radical phosphorylation from 2016 to 2021" Chemical Synthesis. 1, no.1: 9. http://dx.doi.org/10.20517/cs.2021.07
ACS Style
Liu, J.; Xiao H.Z.; Fu Q.; Yu D.G. Advances in radical phosphorylation from 2016 to 2021. Chem. Synth. 2021, 1, 9. http://dx.doi.org/10.20517/cs.2021.07
About This Article
Copyright

Data & Comments
Data


 Cite This Article 86 clicks
Cite This Article 86 clicks

 Like This Article 27
likes
Like This Article 27
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.